David R. Slochower 0000-0003-3928-5050 · slochower · drslochower Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California, San Diego, La Jolla, CA 92093, USA
Niel M. Henriksen 0000-0002-7916-0757 · nhenriksen Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California, San Diego, La Jolla, CA 92093, USA
Lee-Ping Wang 0000-0003-3072-9946 · leeping Department of Chemistry, University of California, Davis, CA 95616, USA
John D. Chodera 0000-0003-0542-119X · jchodera · jchodera Computational and Systems Biology Program, Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center, New York, NY 10065
David L. Mobley 0000-0002-1083-5533 · davidlmobley · davidlmobley Department of Pharmaceutical Sciences and Department of Chemistry, University of California, Irvine, CA 92697, USA
Michael K. Gilson 0000-0002-3375-1738 · michaelkgilson Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California, San Diego, La Jolla, CA 92093, USA
Abstract
Designing ligands that bind their target biomolecules with high affinity and specificity is a key step in small-molecule drug discovery, but accurately predicting protein-ligand binding free energies remains challenging. Key sources of errors in the calculations include inadequate sampling of conformational space, ambiguous protonation states, and errors in force fields. Noncovalent complexes between a host molecule with a binding cavity and a drug-like guest molecules have emerged as powerful model systems. As model systems, host-guest complexes reduce many of the errors in more complex protein-ligand binding systems, as their small size greatly facilitates conformational sampling, and one can choose systems that avoid ambiguities in protonation states. These features, combined with their ease of experimental characterization, make host-guest systems ideal model systems to test and ultimately optimize force fields in the context of binding thermodynamics calculations.
The Open Force Field Initiative aims to create a modern, open software infrastructure for automatically generating and assessing force fields using data sets. The first force field to arise out of this effort, named SMIRNOFF99Frosst, has approximately one tenth the number of parameters, in version 1.0.5, compared to typical general small molecule force fields, such as GAFF. Here, we evaluate the accuracy of this initial force field, using free energy calculations of 43 α and β-cyclodextrin host-guest pairs for which experimental thermodynamic data are available, and compare with matched calculations using two versions of GAFF. For all three force fields, we used TIP3P water and AM1-BCC charges. The calculations are performed using the attach-pull-release (APR) method as implemented in the open source package, pAPRika. For binding free energies, the root mean square error of the SMIRNOFF99Frosst calculations relative to experiment is 0.9 [0.7, 1.1] kcal/mol, while the corresponding results for GAFF 1.7 and GAFF 2.1 are 0.9 [0.7, 1.1] kcal/mol and 1.7 [1.5, 1.9] kcal/mol, respectively, with 95% confidence ranges in brackets. These results suggest that SMIRNOFF99Frosst performs competitively with existing small molecule force fields and is a parsimonious starting point for optimization.
Introduction
The accurate prediction of protein-ligand binding free energies is a central goal of computational chemistry, with key applications in early stage drug discovery. However, calculations of protein-ligand binding thermodynamics still involve a number of challenging choices, including the choice of empirical force field, specifying the protonation states of ionizable residues, adding hydrogens and otherwise adjusting the initial protein structure, and positioning the candidate ligand in the binding pocket. Predictions of protein-ligand absolute binding free energies have achieved root mean square errors around 1-2 kcal/mol for “well-behaved” systems [1,2,3], with deviations an order of magnitude larger for some protein families with slow degrees of freedom [4]. Retrospective relative free energy calculations on a series of congeneric ligands, using proprietary methods, have also achieved root mean square errors compared to experiment of around 1 kcal/mol [5,6,7]. However, it is not possible to determine how much of the prediction error can be attributed to each of the decisions made by the modeler, as opposed to accuracy limitations of the force field.
By minimizing the ambiguities involved in modeling protein-ligand complexes, host-guest systems offer a way to isolate and directly probe force field error. A variety of techniques for computing absolute binding free energies have been applied to host-guest systems, and some have shown accuracy as good as ~1 kcal/mol, as highlighted in the recent SAMPL5 and SAMPL6 blind challenges [1,8]. The techniques applied to this problem have included both quantum and classical dynamics, employing a range of energy and solvation models, with some techniques having knowledge-based steps, docking, or clustering [9,10,11,12,13,14,15,16]. The attach-pull-release (APR) method has consistently been ranked among the most reliable techniques for predicting binding thermodynamics of host-guest complexes in blind challenges [8,17]. In APR, the reversible work of transferring the guest from the binding site to solution, via a physical pathway, is computed using a series of umbrella sampling windows. Simulating each window and integrating over the partial derivative of the restraint energy with respect to the restraint target, in each window, is used to generate a potential of mean force along the pulling coordinate, yielding the binding free energy at standard state, ΔG° after applying an analytic correction to account for the effective concentration of the guest during the simulation [18]. Furthermore, subtracting the mean potential energies obtained from long simulations of the solvated bound complex and the solvated dissociated complex yields the binding enthalpy, ΔH [19]. Together, ΔG° and ΔH can be combined to determine the binding entropy at standard state, ΔS°. Thus, APR provides the complete thermodynamic signature of a host-guest binding reaction: ΔG°, ΔH, and −TΔS°.
Cyclodextrins, in particular, are ideal host molecules for testing computational methods. They are neutral across a broad pH range, with well-characterized structures [20], and bind both small molecule fragments and drug-like guest molecules with reasonable affinity, from near -1 kcal/mol to about -5 kcal/mol in the present work [21], and with higher affinity for some cyclodextrin derivatives [21]. Moreover, cyclodextrins are stable in a wide range of experimental conditions and their high millimolar aqueous solubility allows a range of different experimental techniques to be used to measure their binding to guests [22]. Here, we report the calculation of binding free energies, enthalpies, and entropies of small guest molecules with functional groups often found in drugs to α- and β-cyclodextrin host molecules, converged to within 0.1 kcal/mol statistical uncertainty, using the APR method. These calculations offer an opportunity to benchmark—and ultimately optimize—new and existing force fields.
The first force field produced by the Open Force Field Initiative, SMIRNOFF99Frosst v1.0.5, was released in late 2018 [23,24]. It is derived from AMBER parm99 [25] and Merck’s parm@Frosst [26]. Instead of relying on atom types to assign force field parameters to compounds, which is the procedure followed by the LEaP program used to assign parameters to molecules in AmberTools [27], SMIRNOFF99Frosst and the Open Force Field Toolkit use separately defined local chemical environments for each atom, bond, angle, and dihedral, to apply force field parameters specified by SMIRKS strings [28]. This process simplifies and effectively uncouples the parameters for each term in the force field. For example, the addition of a new Lennard-Jones parameter does not require creating a new atom type that forces the addition of new bonded, angle, and dihedral parameters. This approach leads to a much leaner force field specification; there are over 3000 lines of parameters in GAFF v1.7 [29], over 6000 lines of parameters in GAFF v2.1, and just 322 lines of parameters in SMIRNOFF99Frosst v1.0.5 [30]. It is important to note that SMIRNOFF99Frosst is not yet optimized at this stage, only compressed; subsequent work will focus on optimizing SMIRNOFF99Frosst and other SMIRNOFF-family force fields to fit quantum and experimental data [31]. In the following text, SMIRNOFF99Frosst refers to version 1.0.5 of the force field, unless otherwise noted.
Thus far, SMIRNOFF99Frosst has been tested on hydration free energies of 642 small molecules and the densities and dielectric constants of 45 pure organic liquids [23]. Here, we benchmark SMIRNOFF99Frosst, GAFF v1.7, and GAFF v2.1 using noncovalent binding thermodynamics for 43 host-guest complexes (including two hosts and 33 unique guests) for which experimental thermodynamics data are available, representing three different functional group moieties. We first compare the results of SMIRNOFF99Frosst with those of the conventional force fields GAFF v1.7 and GAFF v2.1, based on calculations of experimental binding free energies, enthalpies, and entropies. We then characterize the differences in host conformations sampled by SMIRNOFF99Frosst compared to the other two force fields.
Methods
Choice of host-guest systems
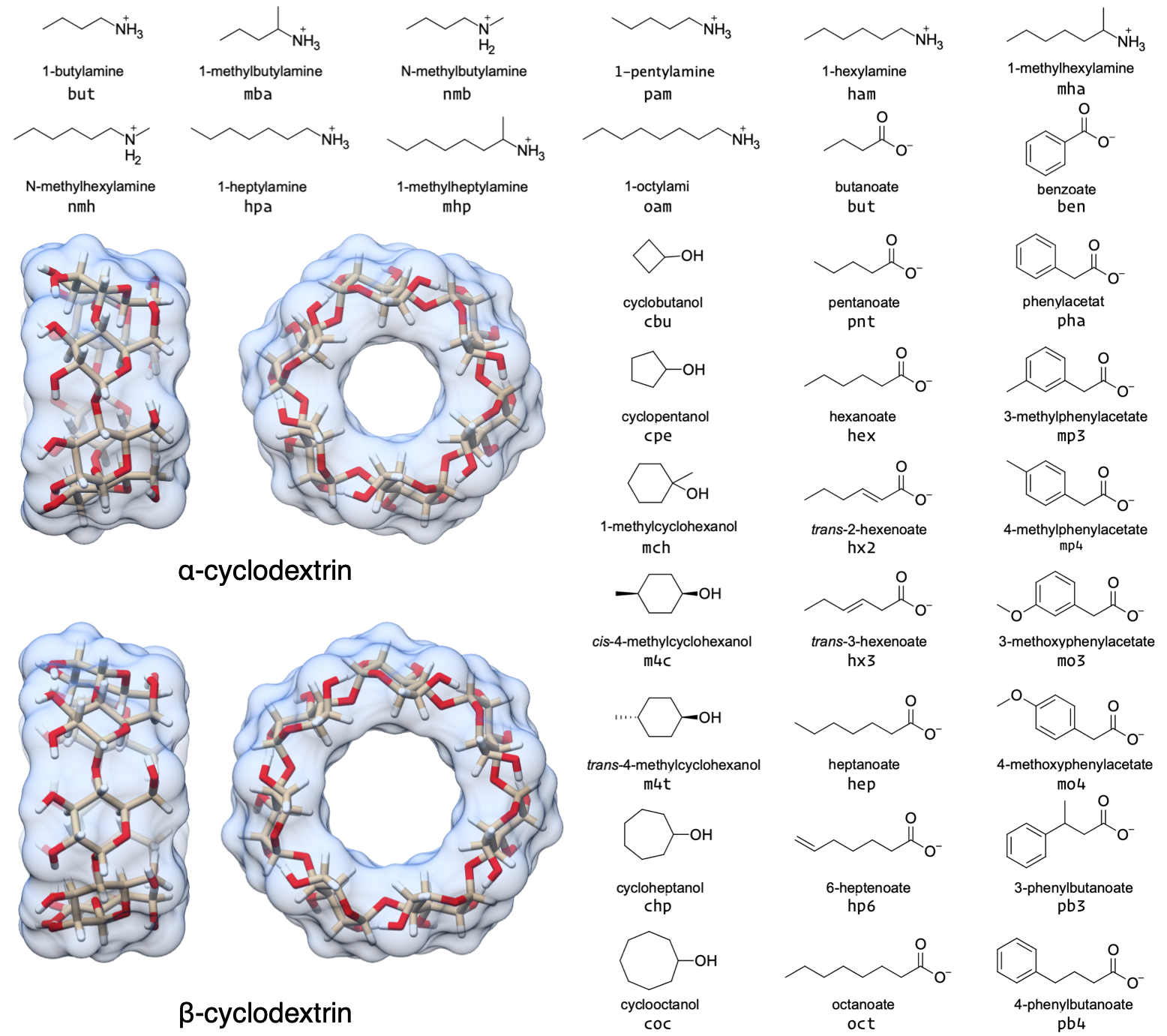
In this study, we report the binding thermodynamics of 43 host-guest complexes (Figure 1 and Table 1) computed using three different force fields. The complexes consist of either α- or β-cyclodextrin as host molecules and a series of small molecule guests containing ammonium, carboxylate, or cyclic alcohol functional groups. The cyclodextrins in the current study are cyclic polymers consisting of six (αCD) or seven (βCD) glucose monomers in the shape of a truncated cone. The equilibrium constants and standard molar enthalpies of binding for these 43 complexes have been measured using isothermal titration calorimetry (ITC) at pH = 6.90 and T = 298 K, and nuclear magnetic resonance spectroscopy (NMR) at pH = 7.0 and T = 298 ± 1 K [32]. Calculations on these host-guest systems have been performed previously [33], and, as in the prior study, we considered only a single stereoisomer for the 1-methylammonium guests because it was not clear whether a mixture or a pure solution was used in Rekharsky, et al. [32], and the ΔG° difference between each stereoisomer is expected to be < 0.1 kcal/mol [34].
Figure 1: Structures of the two cyclodextrin hosts and 33 guest molecules in this study which together comprise 43 unique host-guest pairs. The simulation “residue name” is written beneath each guest.
Table 1: The 43 unique host-guest combinations used in this study. The formal charge of each guest is listed in brackets. The guest names correspond to Tables 1 and 2 in Rekharsky et al. [32]. a Only the R enantiomer was considered. b Only the S enantiomer was considered. SMILES strings are written as canonical isomeric SMILES as implemented in the OpenEye OEChem Toolkit version 2.0.2 [35].
Host-guest ID
Host
Guest
Charge
SMILES
a-bam
αCD
1-butylamine
+1
CCCC[NH3+]
a-nmb
αCD
n-methylbutylamine
+1
CCCC[NH2+]C
a-mba
αCD
1-methylbutylaminea
+1
CCC[C@@H](C)[NH3+]
a-pam
αCD
1-pentylamine
+1
CCCCC[NH3+]
a-ham
αCD
1-hexylamine
+1
CCCCCC[NH3+]
a-nmh
αCD
n-methylhexylamine
+1
CCCCCC[NH2+]C
a-mha
αCD
1-methylhexylaminea
+1
CCCCC[C@@H](C)[NH3+]
a-hpa
αCD
1-heptylamine
+1
CCCCCCC[NH3+]
a-mhp
αCD
1-methylheptylamineb
+1
CCCCCC[C@H](C)[NH3+]
a-oam
αCD
1-octylamine
+1
CCCCCCCC[NH3+]
b-ham
βCD
1-hexylamine
+1
CCCCCC[NH3+]
b-mha
βCD
1-methylhexylaminea
+1
CCCCC[C@@H](C)[NH3+]
b-oam
βCD
1-octylamine
+1
CCCCCCCC[NH3+]
a-cbu
αCD
cyclobutanol
0
C1CC(C1)O
a-cpe
αCD
cyclopentanol
0
C1CCC(C1)O
a-chp
αCD
cycloheptanol
0
C1CCCC(CC1)O
a-coc
αCD
cyclooctanol
0
C1CCCC(CCC1)O
b-cbu
βCD
cyclobutanol
0
C1CC(C1)O
b-cpe
βCD
cyclopentanol
0
C1CCC(C1)O
b-mch
βCD
1-methylcyclohexanol
0
CC1(CCCCC1)O
b-m4c
βCD
cis-4-methylcyclohexanol
0
CC1CCC(CC1)O
b-m4t
βCD
trans-4-methylcyclohexanol
0
CC1CCC(CC1)O
b-chp
βCD
cycloheptanol
0
C1CCCC(CC1)O
b-coc
βCD
cyclooctanol
0
C1CCCC(CCC1)O
a-but
αCD
butanoate
-1
CCCC(=O)[O-]
a-pnt
αCD
pentanoate
-1
CCCCC(=O)[O-]
a-hex
αCD
hexanoate
-1
CCCCCC(=O)[O-]
a-hx2
αCD
trans-2-hexenoate
-1
CCC/C=C/C(=O)[O-]
a-hx3
αCD
trans-3-hexenoate
-1
CC/C=C/CC(=O)[O-]
a-hep
αCD
heptanoate
-1
CCCCCCC(=O)[O-]
a-hp6
αCD
6-heptenoate
-1
C=CCCCCC(=O)[O-]
a-oct
αCD
octanoate
-1
CCCCCCCC(=O)[O-]
b-pnt
βCD
pentanoate
-1
CCCCC(=O)[O-]
b-hex
βCD
hexanoate
-1
CCCCCC(=O)[O-]
b-hep
βCD
heptanoate
-1
CCCCCCC(=O)[O-]
b-ben
βCD
benzoate
-1
c1ccc(cc1)C(=O)[O-]
b-pha
βCD
phenylacetate
-1
c1ccc(cc1)CC(=O)[O-]
b-mp3
βCD
3-methylphenylacetate
-1
Cc1cccc(c1)CC(=O)[O-]
b-mp4
βCD
4-methylphenylacetate
-1
Cc1ccc(cc1)CC(=O)[O-]
b-mo3
βCD
3-methoxyphenylacetate
-1
COc1cccc(c1)CC(=O)[O-]
b-mo4
βCD
4-methoxyphenylacetate
-1
COc1ccc(cc1)CC(=O)[O-]
b-pb3
βCD
3-phenylbutanoate
-1
C[C@H](CC(=O)[O-])c1ccccc1
b-pb4
βCD
4-phenylbutanoate
-1
c1ccc(cc1)CCCC(=O)[O-]
Application of force field parameters
We sought to compare force fields directly and therefore attempted to minimize additional differences among the simulations with each force field. In all simulations, we applied AM1-BCC [36,37] partial atomic charges to both the host and guest molecules using the antechamber program in AmberTools16 [27].
The Open Force Field Toolkit provides a mechanism for user-specified charges. If no charges are supplied, the toolkit will generate AM1-BCC charges. AM1-BCC is the recommended charge scehme, and the
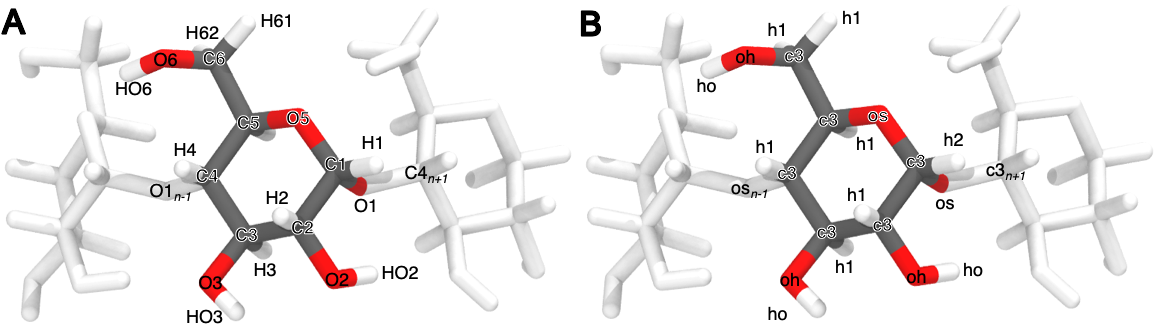
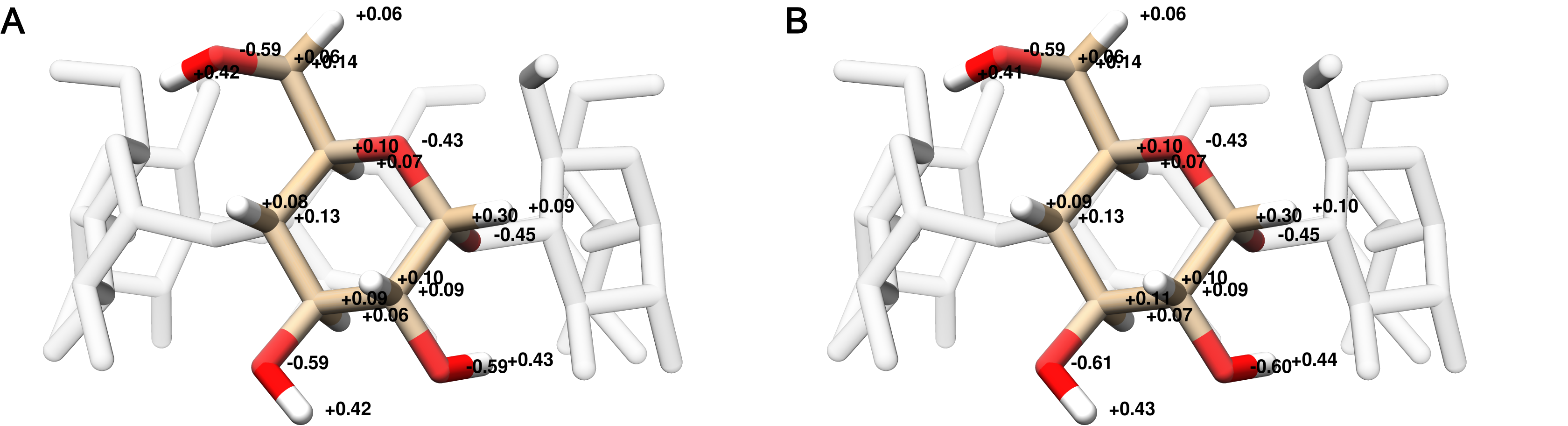
The host charges were calculated using a single glucose molecule with methoxy caps on the O1 and O4 alcohols (Figure 2); each glucose monomer in the cyclodextrin polymer has identical charges. After removing the capping atoms, the net charge of the glucose monomer was -0.064 e. To ensure a neutrality of the glucose monomer, the charge remainder was proportionally distributed across all atoms according to the magnitude of the partial charge for each atom. The minimum and maximum charge adjustments were 0.000684 and 0.007245 e, respectively. Using the entire αCD molecule as an input to antechamber results in partial atomic charges that differ by at most 0.02 e, compared to using a single monomer, and requires reducing the maximum path length used to determine the equivalence of atomic charges (Figure 16). We used TIP3P water [38] and Joung-Cheatham monovalent ion parameters [39] in each simulation set.
Figure 2: Atom names (A) and GAFF atom types (B) for a glucose monomer in αCD shown with two flanking monomers. The remaining three glucose monomers are hidden for clarity.
GAFF v1.7 bond, angle, torsion, and Lennard-Jones parameters were applied using the tleap program distributed with AmberTools16. GAFF v2.1 parameters were applied in an identical manner to the GAFF v1.7 parameters, using the tleap program distributed with AmberTools18 and substituting leaprc.gaff for leaprc.gaff2 in the tleap input file.
To apply SMIRNOFF99Frosst parameters, we followed a multistep process, beginning with the AMBER-format .prmtop and .inpcrd GAFF v1.7 files. The host and guest molecules were parameterized with version 0.0.3 of the Open Force Field Toolkit which uses the OpenEye OEChem Toolkit version 2.0.2 [35], which reads molecular coordinates and topologies and creates a serialized representation of the molecular system; version 1.0.5 of the SMIRNOFF99Frosst force field; specified in version 1.0 of the SMIRNOFF format. Once parameterized with SMIRNOFF99Frosst, the topology and coordinates for the host-guest complex were combined with the solvent and ions, which retained their TIP3P water parameters and Joung-Cheatham ion parameters, respectively. This was accomplished by the ParmEd program [40], which enables saving the OpenMM system created by the Open Force Field Toolkit in AMBER-format .prmtop and .inpcrd files. Ongoing updates to the Open Force Field Toolkit may result in changes to how this procedure is carried out in the future.
Thermodynamic calculation
We used the attach-pull-release (APR) method, as implemented in the open source package pAPRika version 0.0.3, to calculate absolute binding free energies. A complete description of the APR method has been provided in the literature [13,17,19,41]. The attachment and release phases each consisted of 15 independent windows. During the attachment phase, the force constants on the host and guest are scaled by a \(\lambda\) parameter that goes from \(\lambda = 0\), at which point all restraints are turned off, to \(\lambda = 1\), at which point all restraints are at their maximum force constant. The \(\lambda\) windows are more densely spaced where the force constant is smaller to improve sampling along highly curved regions of the potential of mean force. These restraints include a set of distance, angle, and torsion restraints that orient the host and guest along the long axis of the simulation box. A separate set of conformational restraints were applied between neighboring glucose units of the cyclodextrin to minimize deformations of the host molecule as the guest molecule is pulled out. The conformational restraints were applied along the pseudodihedrals O5n–C1n–O1n–C4n+1 and C1n–O1n–C4n+1–C5n+1 to improve convergence and sampling of the bound state (Figure 2 for atom names). To further improve sampling of weak-binding guests, we applied a hard wall restraint that confined the guest molecule to within a sphere of 12.3 and 13.5 Å of αCD and βCD, respectively, during the bound state.
The release phase is the conceptual reverse of the attach phase, in which the conformational restraints on the host are gradually turned off (\(\lambda =1 \rightarrow 0\)) in the absence of the guest. This explicit release phase is performed once for αCD and once for βCD, as it is independent of guest molecule. Finally, an analytic correction is performed to compute the work of moving the guest from the restricted volume enforced by the APR restraints to standard state at 1 M concentration.
The pulling phase consisted of 45 independent, equally spaced windows. During the pulling phase, the \(\lambda\) parameter represents the target value of a distance restraint with constant force constant. This target distance is increased uniformly in 44 increments of 0.4 Å, yielding windows that separate the host and guest by 18 Å over the course of the calculation.
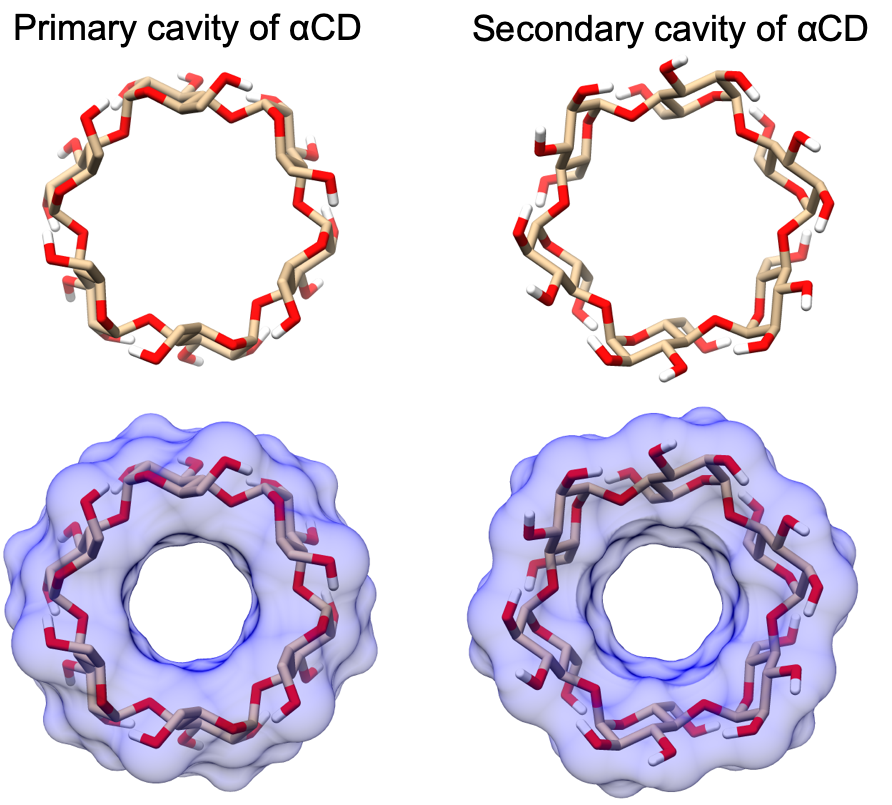
Due to the asymmetry of the primary and secondary alcohols of cyclodextrin (Figure 18), as well as of the small molecule guests, there are generally two distinct binding poses that do not interconvert during the simulation timescale. To account for this effect, we separately compute the binding free energy and enthalpy for each orientation [13] and combine the results to produce a single value for each host-guest combination using the following equation:
In this manuscript, we refer to calculations where the guest functional group in the bound state is at the primary face of cyclodextrin with a -p suffix, and calculations where it is at the secondary face of cyclodextrin with a -s suffix..
Thermodynamic integration [42] and the multistate Bennett acceptance ratio estimator (MBAR) [43] were used to compute the binding free energy (ΔG°). The results presented in the main text are those analyzed using thermodynamic integration to be consistent with prior analysis presented in Henriksen, et al.[33]. The binding enthalpy (ΔH) was computed as the difference in mean potential energy of the bound state (in the absence of any restraints) and the unbound state (where the guest is held far away from the host, but the conformational restraints on the host are disabled). The binding entropy (ΔS°) was computed by subtraction using ΔG° and ΔH.
Simulations
Simulations were performed with the pmemd.cuda module of AMBER 16 (calculations with the GAFF v1.7 force field) and AMBER 18 (calculations with the GAFF v2.1 and SMIRNOFF99Frosst force fields) molecular dynamics software [27,44]. Each window for each system was independently solvated and simulated. Simulation data for the host-guest complexes using GAFF v1.7 were taken from Henriksen, et al.[33] and are described in additional detail therein. Solvation consisted of 2000 TIP3P waters for the αCD systems and 2210 waters for the βCD systems in an orthorhombic box. The host and guest were oriented via non-interacting dummy atoms along the simulation box’s long \(z\) axis, to allow use of an elongated periodic box that reduces the amount of solvent required for the calculation. Each simulation contained enough Na+ or Cl- ions to neutralize the host-guest complex and an additional 50 mM NaCl to match the experimental conditions in [32]. In the GAFF simulations, hydrogen mass repartitioning [45] was used to adjust the mass of hydrogen atoms by a factor of 3 and decreasing the mass of the bound heavy atoms proportionally, keeping the total molecular weight of each molecule constant and enabling a simulation timestep of 4 fs. Hydrogen mass repartitioning produces negligible changes in computed thermodynamic observables for other cyclodextrin-guest calculations, with deviations within statistical uncertainty [13]. Equilibration consisted of 50,000 steps of energy minimization, 100 ps of heating from 0 to 300 K, and then 2000 ps of additional NPT simulation. AMBER’s Langevin thermostat with a collision rate of 1 ps-1, the Monte Carlo barostat, a nonbonded cutoff of 9 Å and default PME parameters, were used for the NPT simulations. An isotropic analytic correction to the Lennard-Jones interactions is applied beyond the cutoff distance [46]. Production NPT simulations were run for a minimum of 2.5 ns and maximum of 50 ns per window, except for the windows used to calculate the enthalpy, which were each simulated for 1 μs. In the GAFF v1.7 and GAFF v2.1 simulations, the exact length of each window’s simulation was determined by the uncertainty in the work done in each λ window. In particular, for restraint energy \(U\) in \(\lambda\) window \(i\), we define the instantaneous SEM of \(\partial U/\partial λ_i\) as σ(λi), and each window (except for the windows used to calculate ΔH) was simulated until the value of \(w(\lambda_i)\), defined as
\[
w(\lambda_i) =
\begin{cases}
\sigma(\lambda_i) \, \frac{\lambda_{i+1}}{2} & i = 0 \\
\sigma(\lambda_i) \, \frac{\lambda_{i+1} - \lambda_{i-1}}{2} & i \in [1, N-1] \\
\sigma(\lambda_i) \, \frac{1 - \lambda_{i-1}}{2} & i = N \\
\end{cases}
\](1)
fell below a threshold of 0.02 kcal/mol during the attach phase and 0.1 kcal/mol during the pull phase.
The second term in Equation 1 scales the uncertainty in the work in each λ window by the nonuniform spacing of the λ windows. \(w(\lambda_i)\) is the approximate contribution of window λi to the overall PMF uncertainty. Excluding the first and last window, the average window length was 11.8 ns and 5.39 ns for GAFF v1.7 and GAFF v2.1 simulations, respectively. We took a more direct approach with the SMIRNOFF99Frosst simulations, due to changes in pAPRika that allowed us to target uncertainties of the same magnitude as in the GAFF simulations, by running each window for a constant length of 10 ns, except for the first and last window which ran for 1 μs to converge ΔH for all three force fields.
Statistics
The uncertainty in the work done by each restraint in each simulation window, σ(λi), was estimated using blocking analysis [47], in a manner which has been shown to yield good agreement with uncertainties obtained from independent replicates [13]. In particular, rather than looking for a plateau in the SEM as the size of the blocks increased, as originally described by Flyvbjerg and Peterssen [47], we instead use the largest standard error of the mean (SEM) obtained for any block size. This avoids the requirement of detecting a plateau and yields a more conservative estimate; i.e., a larger SEM. Then, using Gaussians with the mean and SEM of \(\frac{\partial U}{\partial \lambda}\) in each window, new values of \(\frac{\partial U}{\partial \lambda}\) were bootstrap sampled for each window 100,000 times and combined to create artificial data for 100,000 notional APR calculations. These were integrated across all windows with splines to generate 100,000 estimates of ΔG°. We report the mean and standard deviation of these 100,000 results as the final mean and its SEM. The SEM of ΔH was computed from the SEM of the total potential energy in each end point window, estimated using blocking analysis, added in quadrature. The standard error of the mean of −TΔS° was calculated using the uncertainties in ΔG° and ΔH added in quadrature.
For each force field, we computed the root mean squared error (RMSE), mean signed error (MSE), the coefficient of determination (R2), Kendall’s rank correlation coefficient (τ), and the slope and intercept of the linear regressions of the computed properties against the experimental values. The R2 values for the subsets of ligand with each are also reported in the bottom right corner in each graph. Comparisons with experiment have 43 measurements, for the 43 unique host-guest complexes listed in Table 1; comparisons between force fields have 86 data points, representing the calculations for the two orientations of the guest, “p” and “s”, in the binding site (see above). The overall RMSE and R2 statistics for each comparison are reported as the sample mean estimated from using all the data, with the 95% confidence interval, from bootstrapping over the set of complexes, in brackets.
Results
This results section is organized as follows. We first present a comparison of binding free energies (ΔG°) and binding enthalpies (ΔH) of small molecule guests to α-cyclodextrin (αCD) and β-cyclodextrin (βCD), computed with SMIRNOFF99Frosst and two versions of the General AMBER Force Field (GAFF [29]). We then detail how the conformational preferences of the host molecules changes between force fields and seek insight into key parameter differences between SMIRNOFF99Frosst and GAFF and their effects.
Comparison with experimental binding free energies, enthalpies, and entropies
Binding free energies
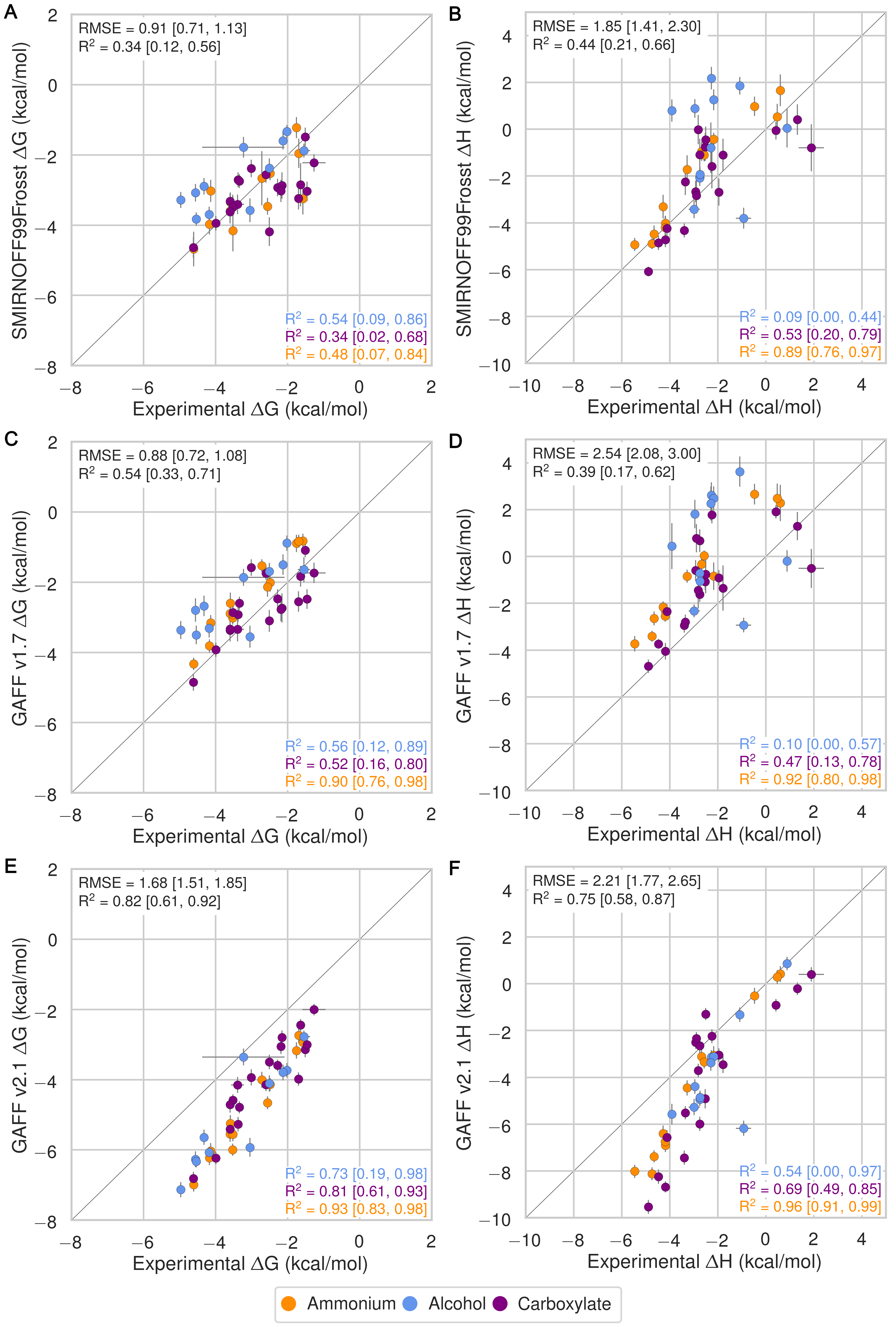
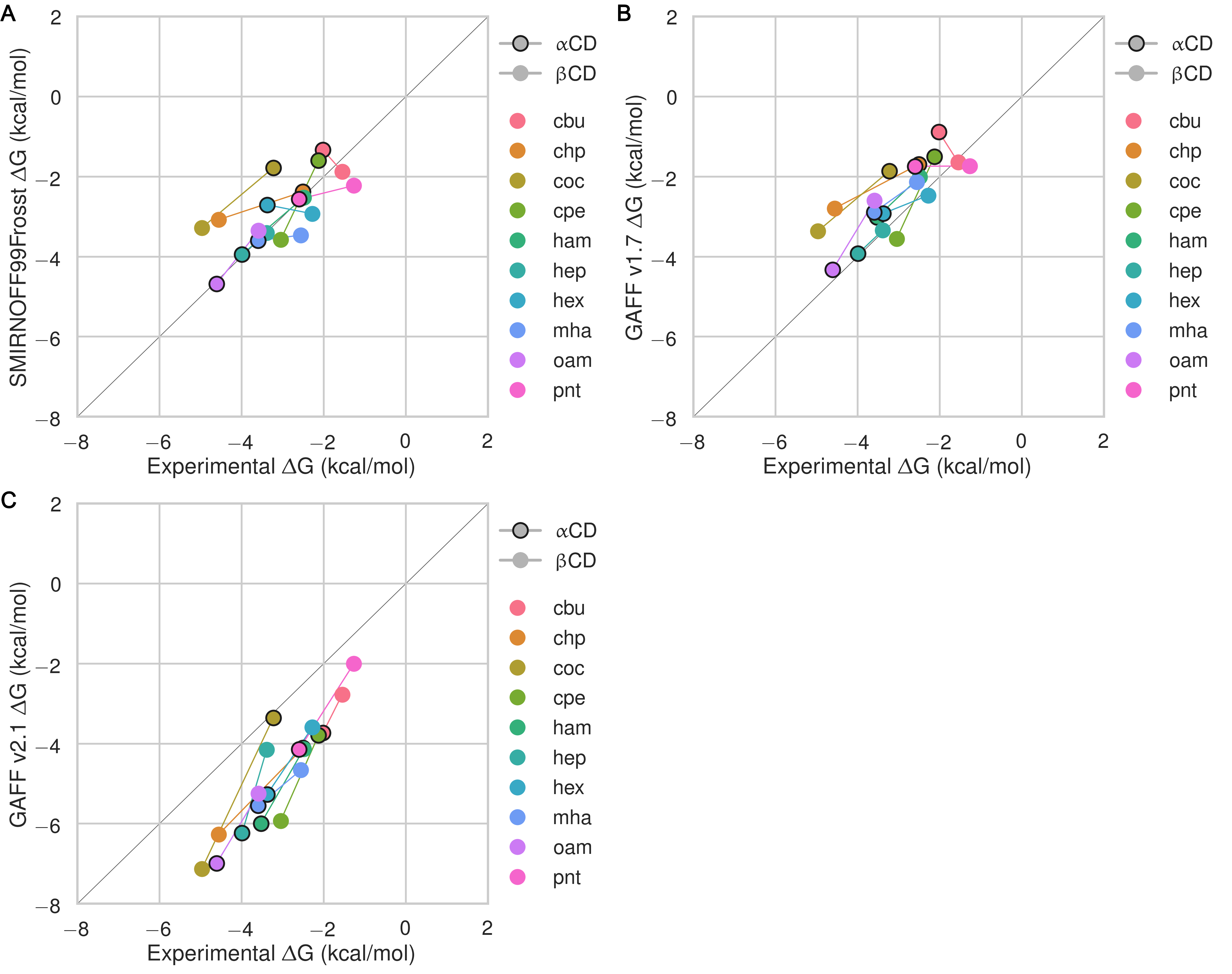
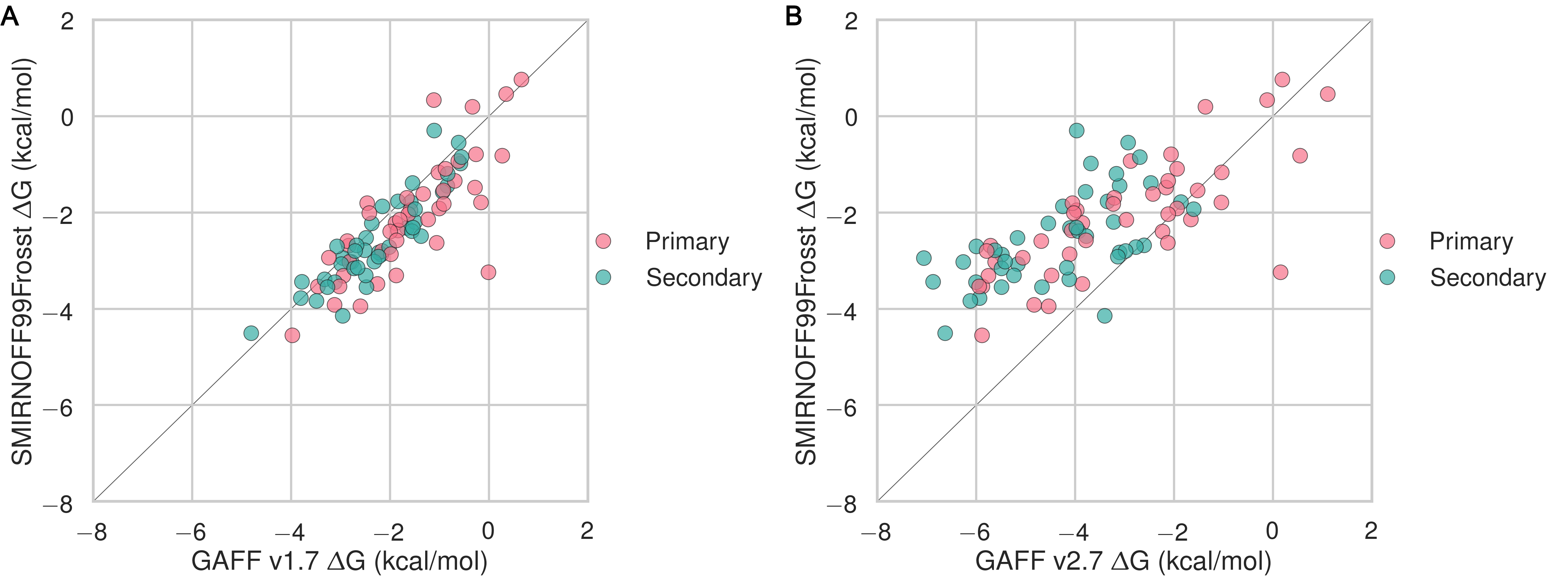
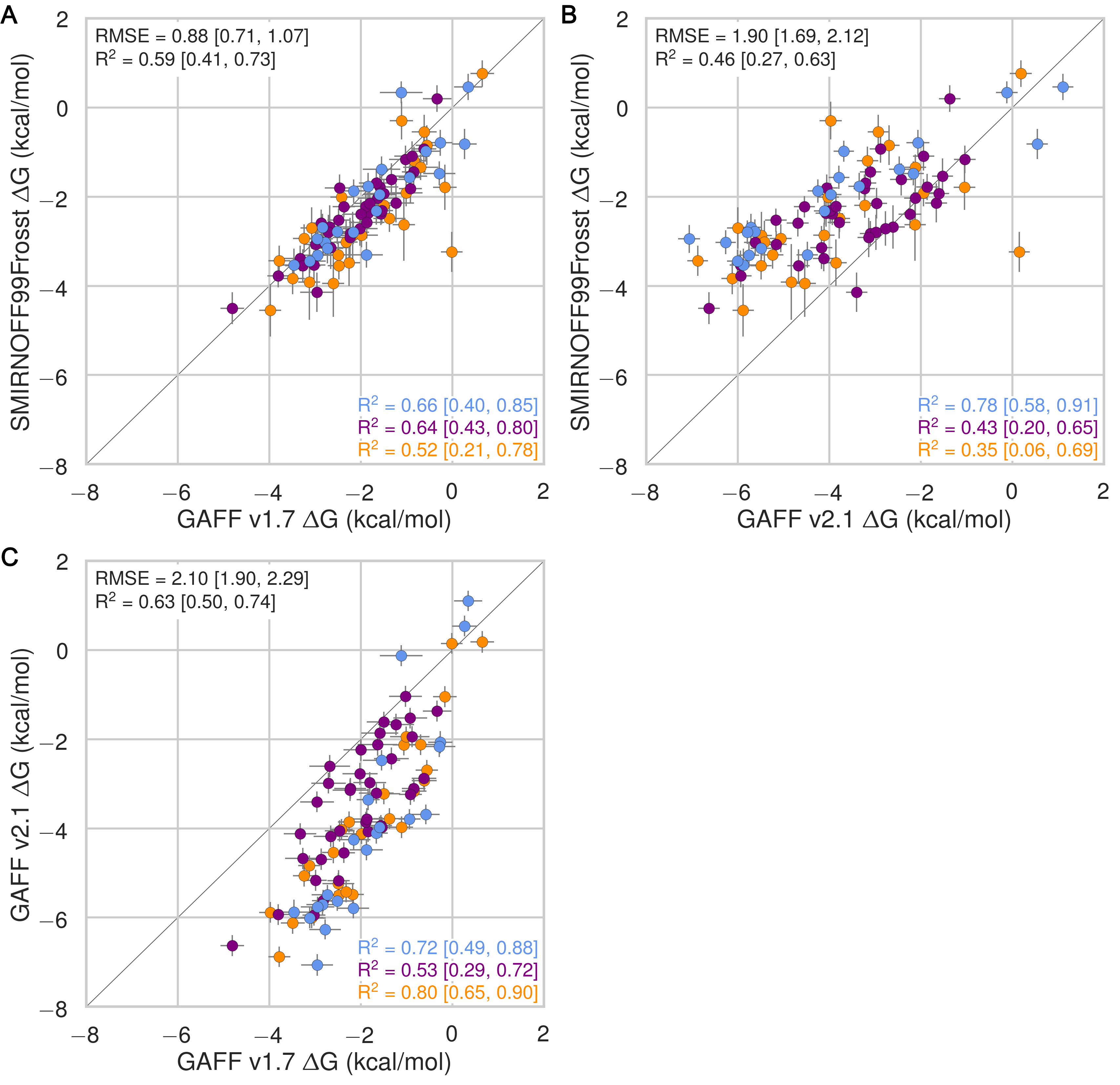
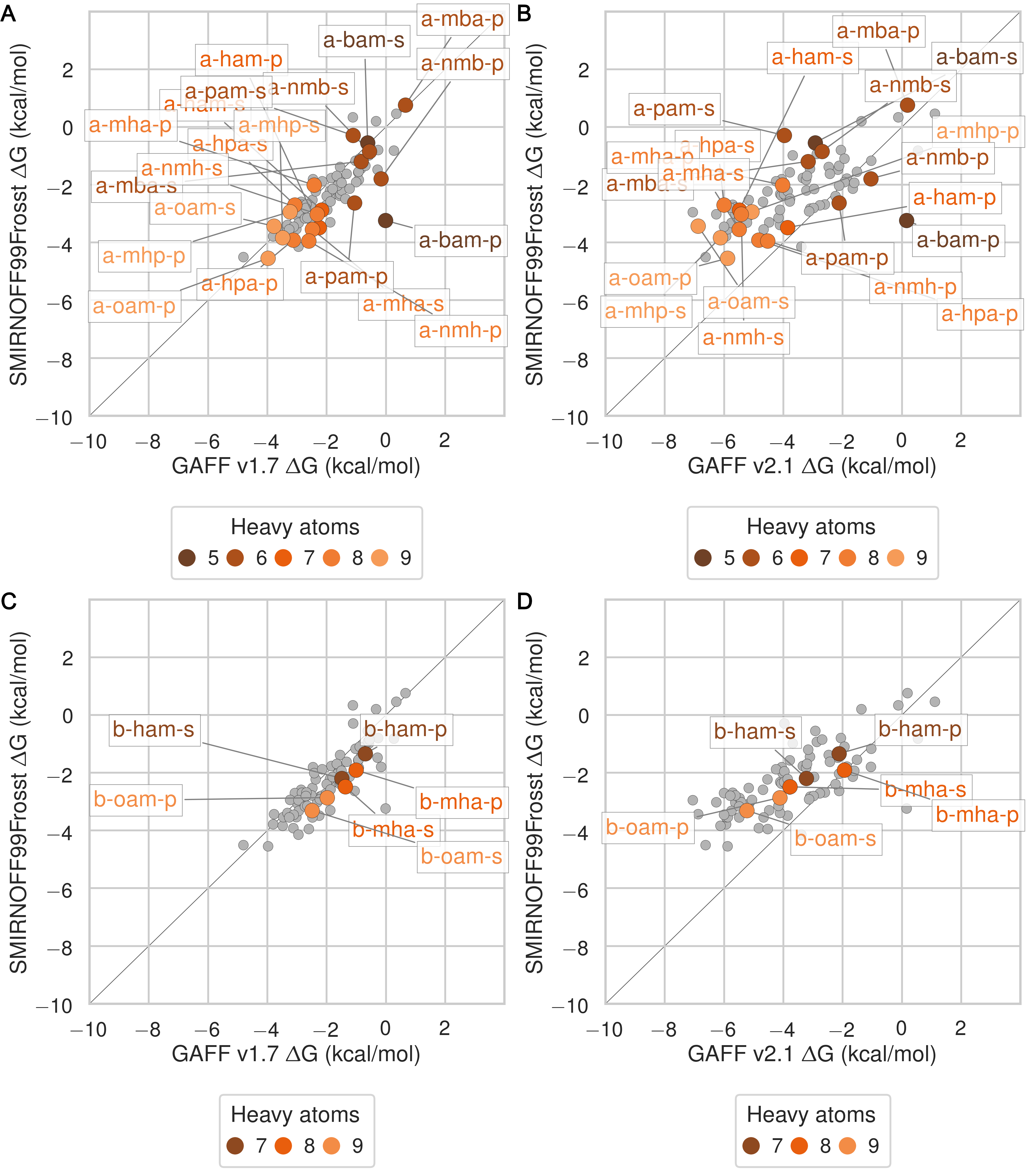
Despite having far fewer numerical parameters, SMIRNOFF99Frosst does about as well as GAFF v1.7 and arguably better than GAFF v2.1 at replicating binding free energies measured by ITC or NMR. Thus, SMIRNOFF99Frosst yields an overall ΔG° RMSE from experiment of 0.9 [0.7, 1.1] kcal/mol across the 43 host-guest systems, compared to the statistically indistinguishable 0.9 [0.7, 1.1] kcal/mol for GAFF v1.7, and distinct from 1.7 [1.5, 1.9] kcal/mol for GAFF v2.1 (where the 95% confidence interval is written in brackets) as detailed in Figure 3; Tables 2, 5.
On the whole, GAFF v1.7 agrees well with SMIRNOFF99Frosst (Figure 21), as the RMSE and MSE between their results are 0.8 [0.6, 1.0] kcal/mol and -0.5 [-0.3, -0.7] kcal/mol. This result is not surprising as GAFF v1.7 and SMIRNOFF99Frosst may be considered cousin force fields with a common ancestor in AMBER’s parm99. Both SMIRNOFF99Frosst and GAFF v1.7 systematically underestimate the binding affinity for cyclic alcohols, with MSEs of 0.7 [0.2, 1.2] kcal/mol and 0.9 [0.4, 1.4] kcal/mol, respectively. In contrast, GAFF v2.1 significantly overestimates the binding of all compounds, leading to MSE and RMSE values of -1.6 [-1.7, -1.4] kcal/mol and 1.6 [1.4, 1.8] kcal/mol, respectively. However, GAFF v2.1 has a particularly good correlation with experiment across all functional group classes, with R² of 0.8 [0.6, 0.9], compared with 0.3 [0.1, 0.6] and 0.5 [0.3, 0.7] for SMIRNOFF99Frosst and GAFF 1.7, respectively. This may trace to differences in the host conformations sampled by GAFF v2.1, which indicate a more consistently open cyclodextrin “pocket” for guests to bind (Figure 14), as detailed below.
Binding enthalpies and entropies
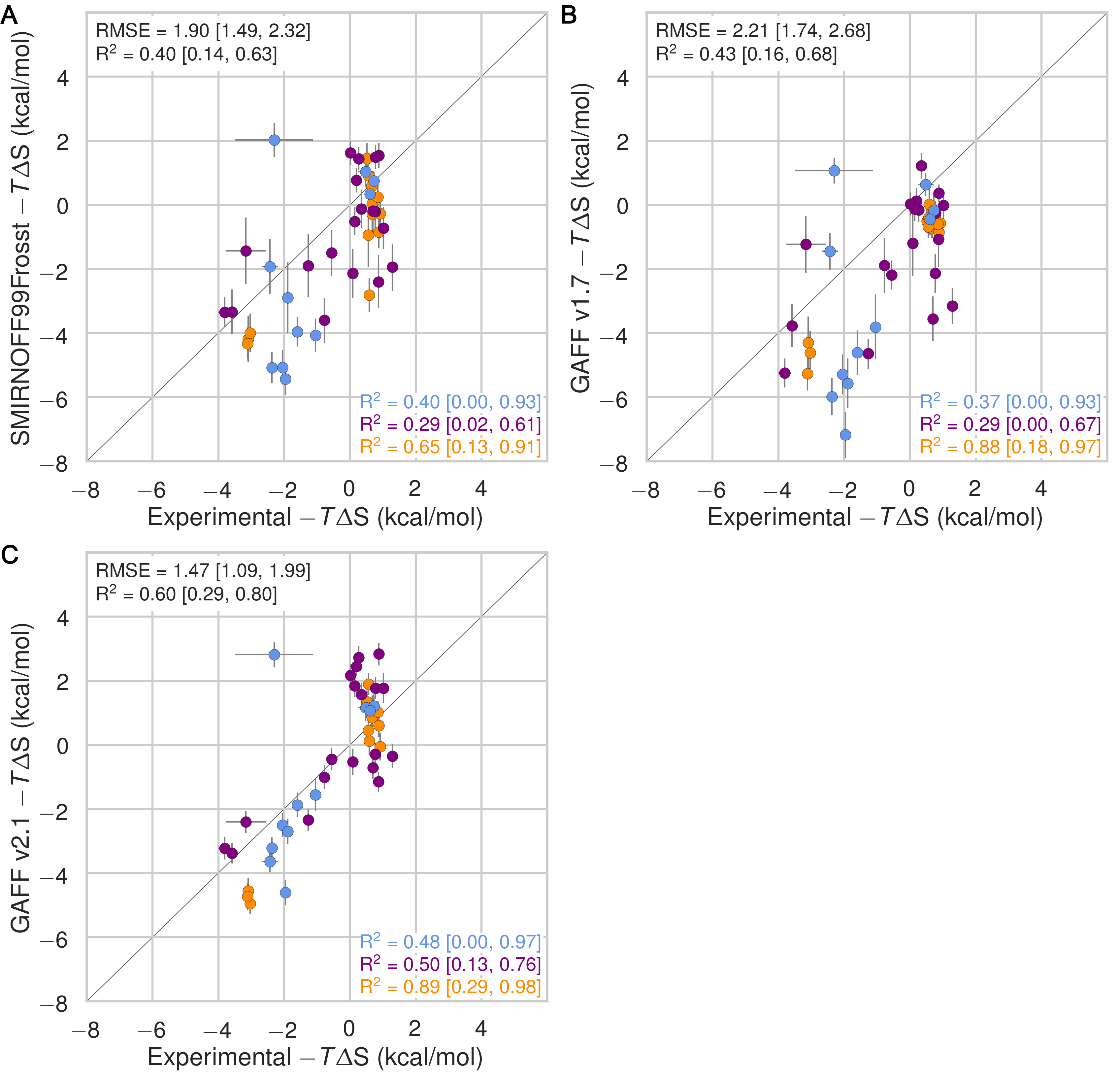
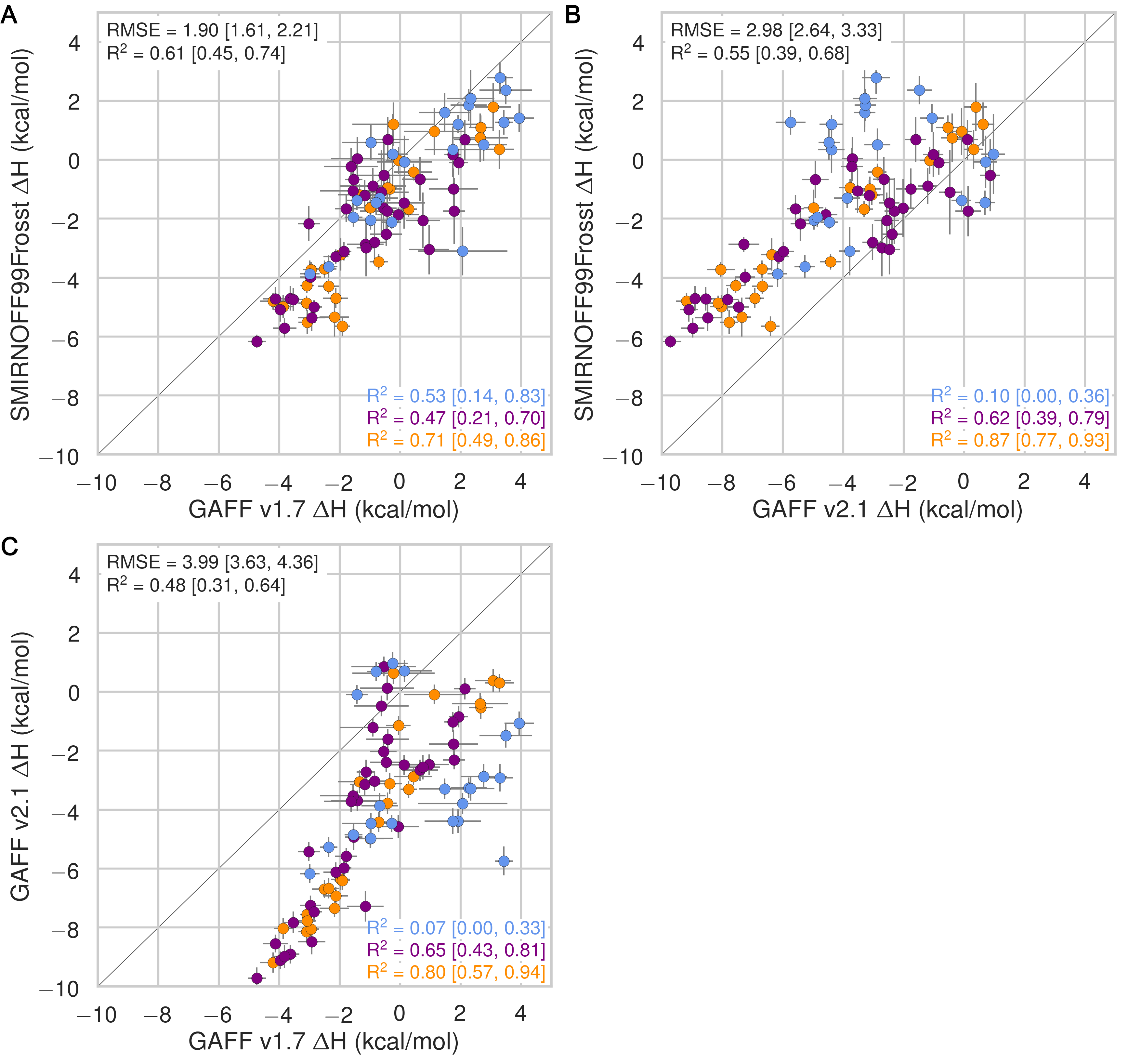
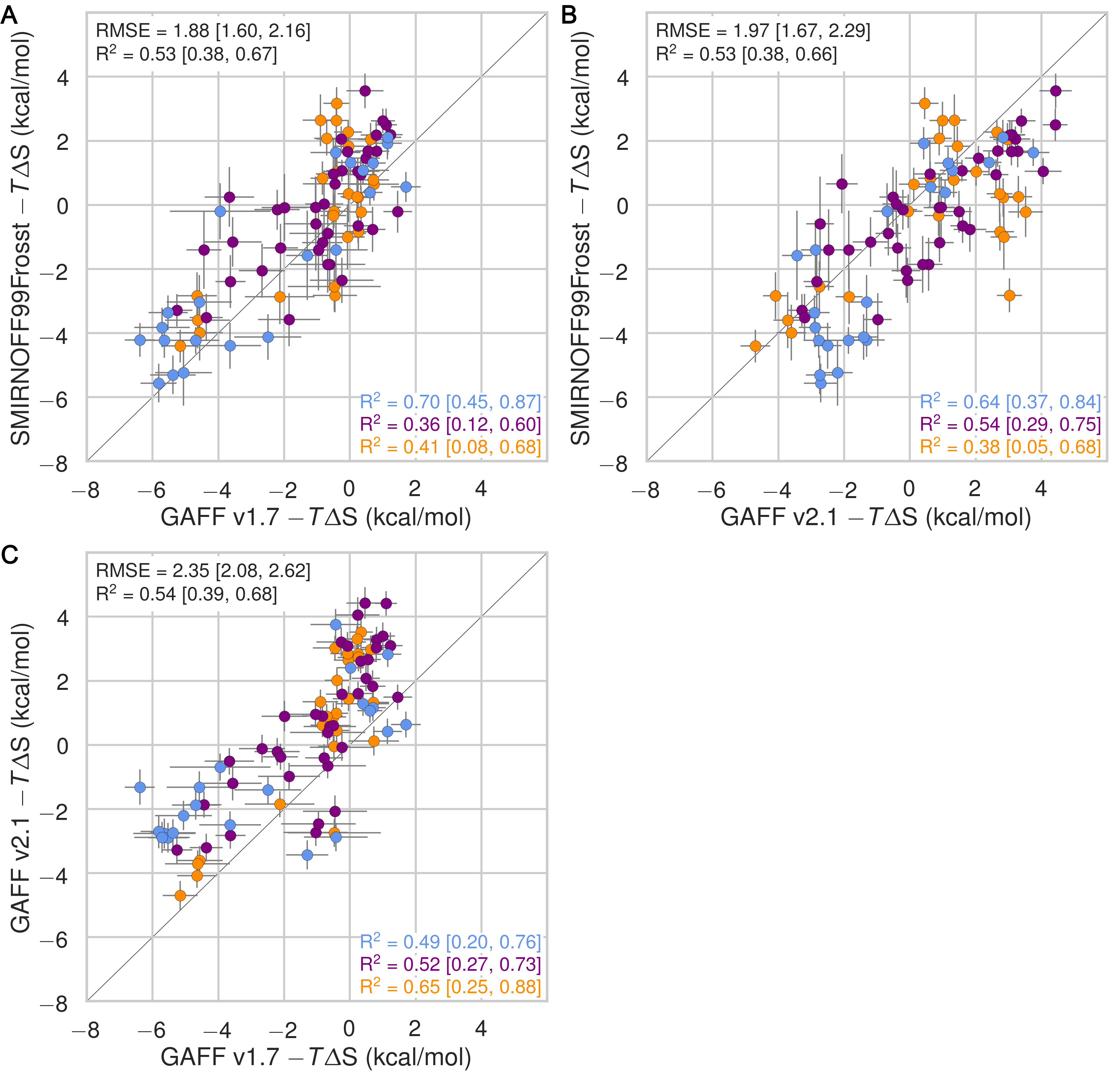
In the case of binding enthalpies (Figure 3), SMIRNOFF99Frosst agrees the best with experiment (RMSE 1.8 [1.4, 2.3] kcal/mol), followed by GAFF v2.1 (RMSE = 2.2 [1.8, 2.7] kcal/mol), and then GAFF v1.7 (RMSE = 2.5 [2.0, 3.0] kcal/mol). In some cases, GAFF v1.7 underestimates ΔH by over 3 kcal/mol and up to 5 kcal/mol (b-chp). For binding entropies, GAFF v2.1 has the lowest RMSE relative to experiment (RMSE = 1.47 [1.1, 2.0] kcal/mol), followed by SMIRNOFF99Frosst (RMSE = 1.9 kcal/mol [1.5, 2.3]), and GAFF v1.7 (RMSE = 2.2 [1.7, 2.7] kcal/mol) (Figure 17).
All force fields perform poorly at replicating −TΔS° for carboxylate guests, with RMSEs ranging from 1.8 [0.7, 3.2] kcal/mol (GAFF v2.1) to 3.0 [2.1, 3.9] kcal/mol (GAFF v1.7). All force fields also underestimate the entropic component of binding of a-coc (αCD:cyclooctanol) relative to experiment, by 3-5 kcal/mol. This is likely due to the poor fit of cycloctanol inside the cavity of αCD, particularly in the primary orientation (Figure 4). Overall, SMIRNOFF99Frosst and GAFF v1.7 yield rather different binding enthalpies (RMSE = 1.6 [1.3, 2.0] kcal/mol) and entropies (RMSE = 1.6 [1.2, 2.0] kcal/mol). The deviations between SMIRNOFF99Frosst and GAFF v2.1 are higher for ΔH (RMSE = 3.0 [2.5, 3.4] kcal/mol) and lower for −TΔS° (RMSE = 1.9 [1.6, 2.2] kcal/mol).
Figure 3: Comparison of calculated absolute binding free energies (ΔG°) and binding enthalpies (ΔH) with experiment with SMIRNOFF99Frosst parameters (A, B), GAFF v1.7 parameters (C, D), or GAFF v2.1 parameters (E, F) applied to both host and guest. The orange, blue, and purple coloring distinguish the functional group of the guest as an ammonium, alcohol, or carboxylate, respectively.
Table 2: Predicted thermodynamic properties for each force field relative to experiment in kcal/mol.
RMSE
MSE
R²
Slope
Intercept
Tau
ΔG°
SMIRNOFF99Frosst
0.91
[0.71, 1.13]
-0.01
[-0.28, 0.26]
0.34
[0.12, 0.56]
0.49
[0.26, 0.72]
-1.55
[-0.80, -2.29]
0.40
[0.57, 0.23]
ΔG°
GAFF v1.7
0.88
[0.72, 1.08]
0.46
[0.23, 0.69]
0.54
[0.33, 0.71]
0.69
[0.47, 0.91]
-0.48
[0.22, -1.16]
0.52
[0.65, 0.38]
ΔG°
GAFF v2.1
1.68
[1.51, 1.85]
-1.56
[-1.74, -1.37]
0.82
[0.61, 0.92]
1.19
[0.96, 1.34]
-1.00
[-0.52, -1.62]
0.73
[0.82, 0.61]
ΔH
SMIRNOFF99Frosst
1.85
[1.41, 2.30]
0.76
[0.26, 1.28]
0.44
[0.21, 0.66]
0.85
[0.54, 1.19]
0.41
[1.55, -0.50]
0.53
[0.69, 0.34]
ΔH
GAFF v1.7
2.54
[2.08, 3.00]
1.84
[1.31, 2.37]
0.39
[0.17, 0.62]
0.80
[0.47, 1.18]
1.36
[2.67, 0.31]
0.50
[0.65, 0.32]
ΔH
GAFF v2.1
2.21
[1.77, 2.65]
-1.64
[-2.10, -1.20]
0.75
[0.58, 0.87]
1.38
[1.15, 1.63]
-0.69
[0.16, -1.43]
0.67
[0.79, 0.52]
−TΔS°
SMIRNOFF99Frosst
1.90
[1.49, 2.32]
-0.78
[-1.29, -0.24]
0.40
[0.14, 0.63]
0.90
[0.51, 1.29]
-0.83
[-0.34, -1.34]
0.33
[0.50, 0.13]
−TΔS°
GAFF v1.7
2.21
[1.74, 2.68]
-1.38
[-1.90, -0.86]
0.43
[0.16, 0.68]
0.95
[0.54, 1.38]
-1.41
[-0.96, -1.89]
0.32
[0.50, 0.10]
−TΔS°
GAFF v2.1
1.80
[0.68, 3.19]
-0.00
[-0.98, 1.27]
0.48
[0.00, 0.97]
1.13
[-0.22, 1.96]
0.08
[1.14, -1.79]
0.46
[0.82, -0.02]
Analysis of the simulations with MBAR produces very slightly improved results for SMIRNOFF99Frosst ΔG°, ΔH, and −TΔS° compared to experiment (Table 8), but they do not appear to be statistically significant.
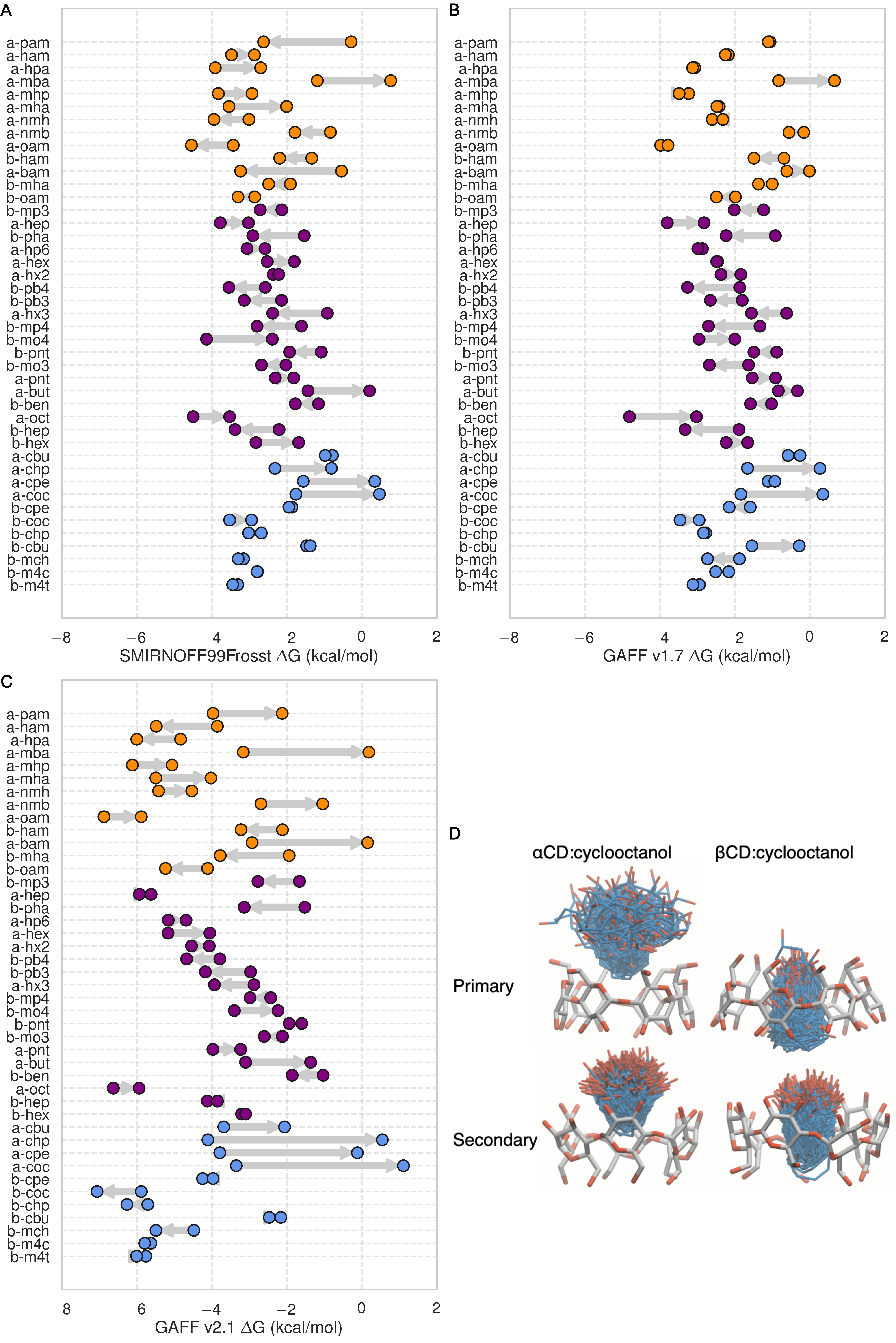
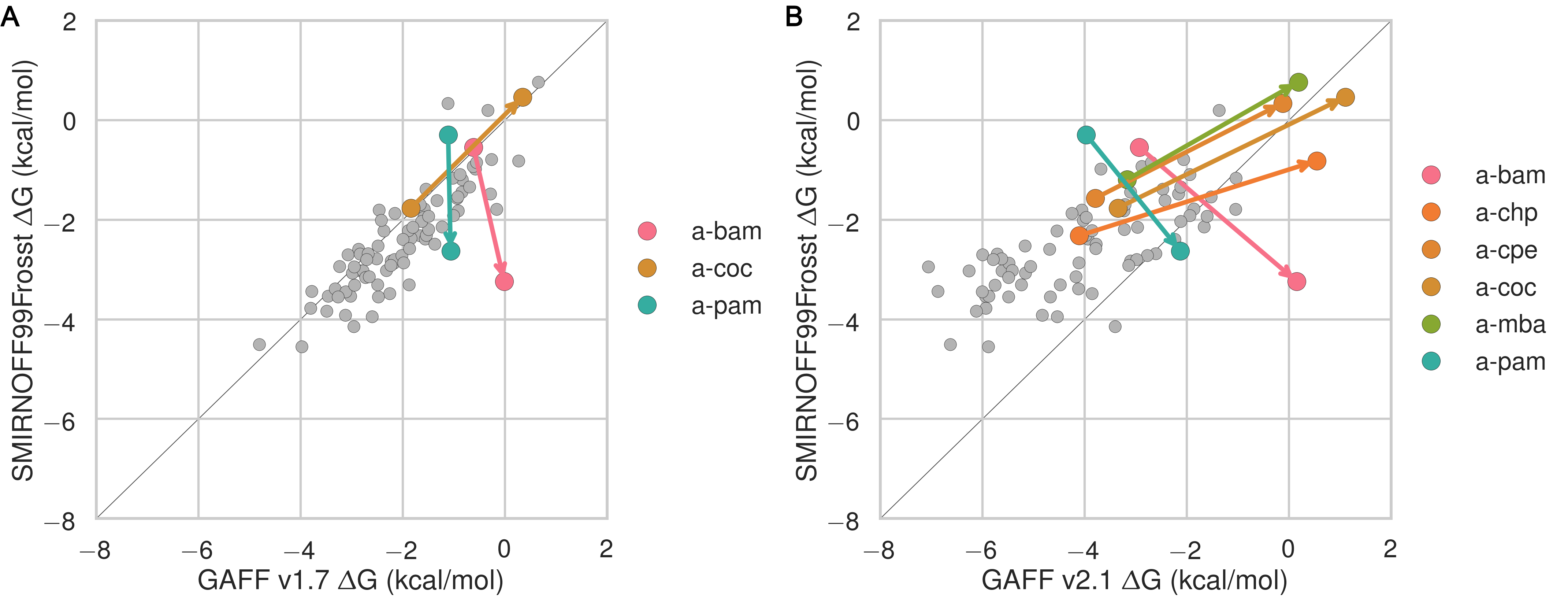
Guest preferences for binding in the primary or secondary orientation
The asymmetry of the hosts and the guests leads to two distinct bound states for each host-guest pair: one where the functional group of the guest sits at the primary face of the host and another where the functional group of the guest sits at the secondary face (18). The difference in binding free energy between these two orientations (ΔΔGorientation) can be large, at around 2 kcal/mol for SMIRNOFF99Frosst and GAFF v1.7 and 5 kcal/mol for GAFF v2.1. SMIRNOFF99Frosst predicts the largest ΔΔGorientation for the ammonium-containing butylamine and pentylamine guests with αCD (4), with the primary orientation being more favorable. Thus, the cationic ammonium groups are predicted to prefer the narrower primary portal of the host. GAFF v1.7 predicts a large ΔΔGorientation for the cyclic alcohols cyclooctanol and cycloheptanol, with the secondary orientation having a more favorable ΔG. When GAFF v2.1 is used, the differences between primary and secondary binding range even higher, greater than 4 kcal/mol, for αCD with these two guests. This effect is due, at least in part, to steric clashes in the bound state for very large guests (Figure 4, D), especially in the narrow primary cavity of the smaller αCD. It is worth noting that the experimental measurement for the the a-coc (αCD:cyclooctanol) complex has very large uncertainties associated with both ΔG° and ΔH.
Figure 4: Comparisons of binding free energy (ΔG) between guests in either the primary or secondary orientation of αCD or βCD, for SMIRNOFF99Frosst (A), GAFF v1.7 (B), or GAFF v2.1 (C). Arrows point from ΔG° for the secondary to ΔG° for the primary cavity. (D) An overlay of cyclooctanol bound state positions (400 snapshots over 1 μs) with αCD (left) or βCD (right) in GAFF v2.1.
Comparison of results for αCD versus βCD
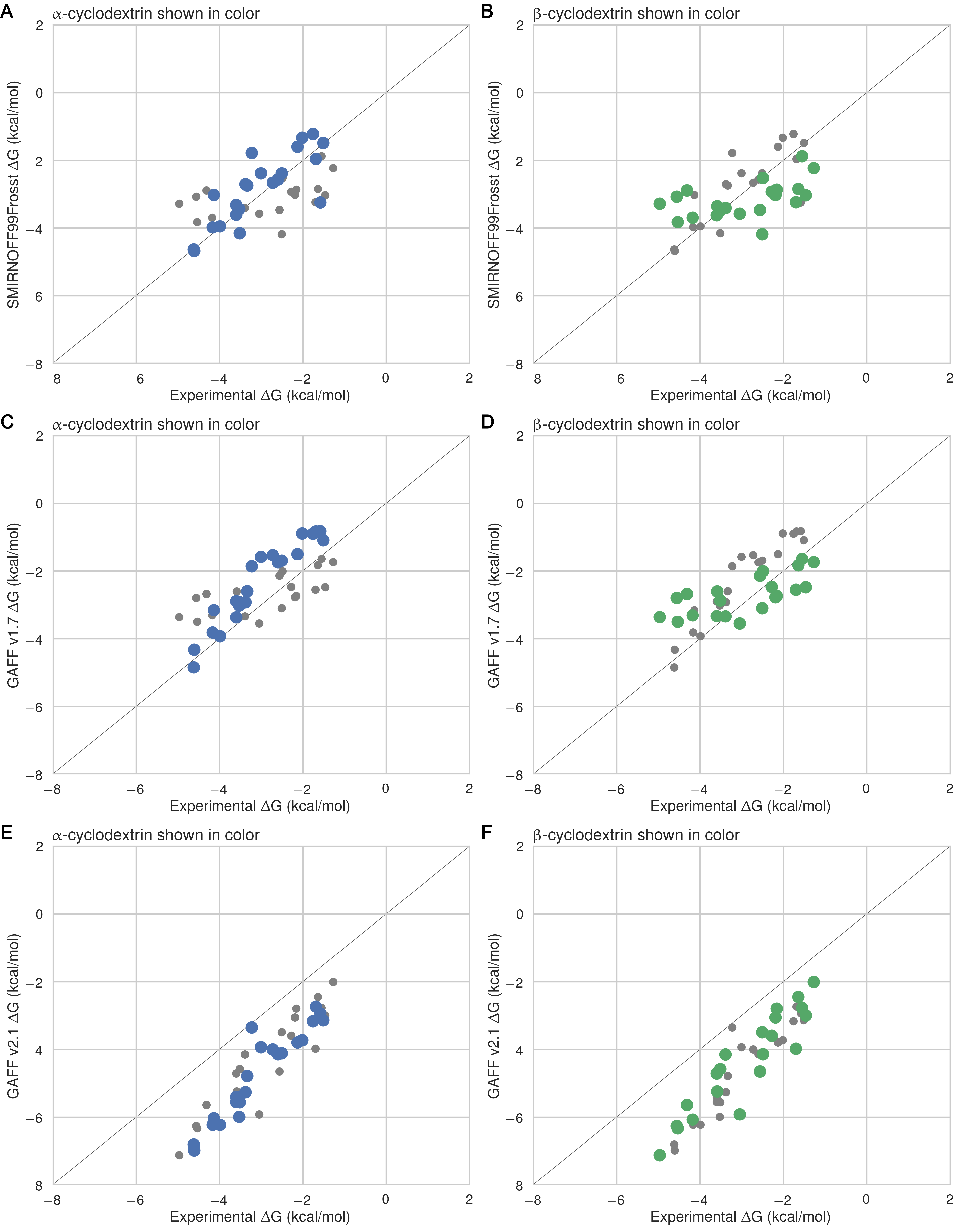
It is of interest to compare the results between αCD and βCD by focusing on the ten guests for which experimental data are available with both hosts. The SMIRNOFF99Frosst and GAFF 1.7 force fields both yield somewhat more accurate binding affinities for αCD (RMSE = 0.8 [0.5, 1.1] kcal/mol) than for βCD (RMSE = 1.0 [0.8, 1.3] kcal/mol), whereas no clear patterns is observed for GAFF v2.1 (Figure 24). Much as seen for the two orientations of the guest molecules within each host, GAFF v2.1 yields relatively large differences in predicted free energies for each guest between the two hosts, but it does not seem to be more accurate for either host relative to the other.
Figure 5: Shown are the αCD and βCD binding free energies for each guest, highlighting the differences in binding to the two hosts for SMIRNOFF99Frosst (A), GAFF v1.7 (B), or GAFF v2.1 (C). The binding affinity for αCD is circled in black. Thin colored lines connect data points for the same guest. Color is used purely to distinguish among the guests.
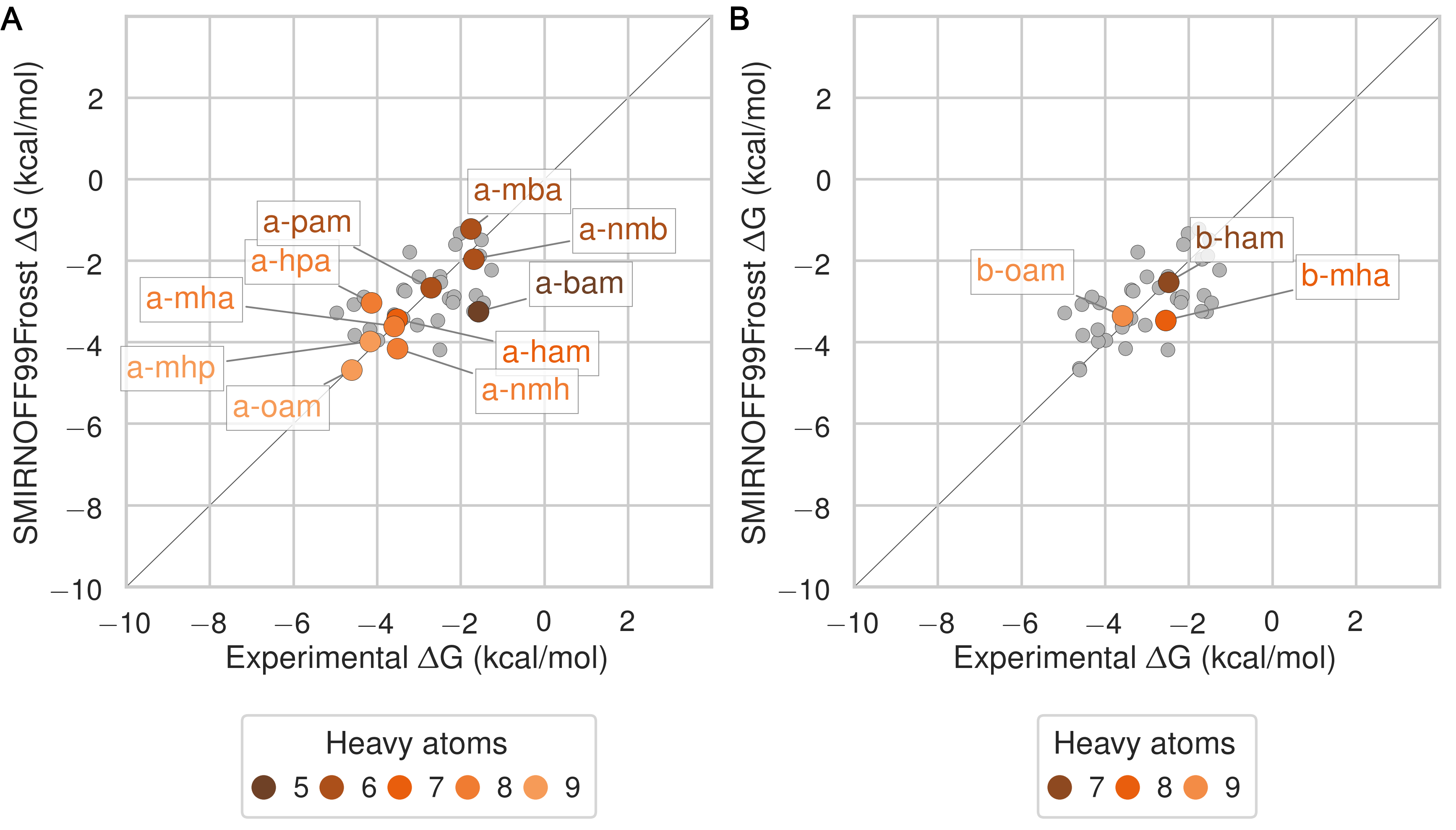
Trends by guest functional group
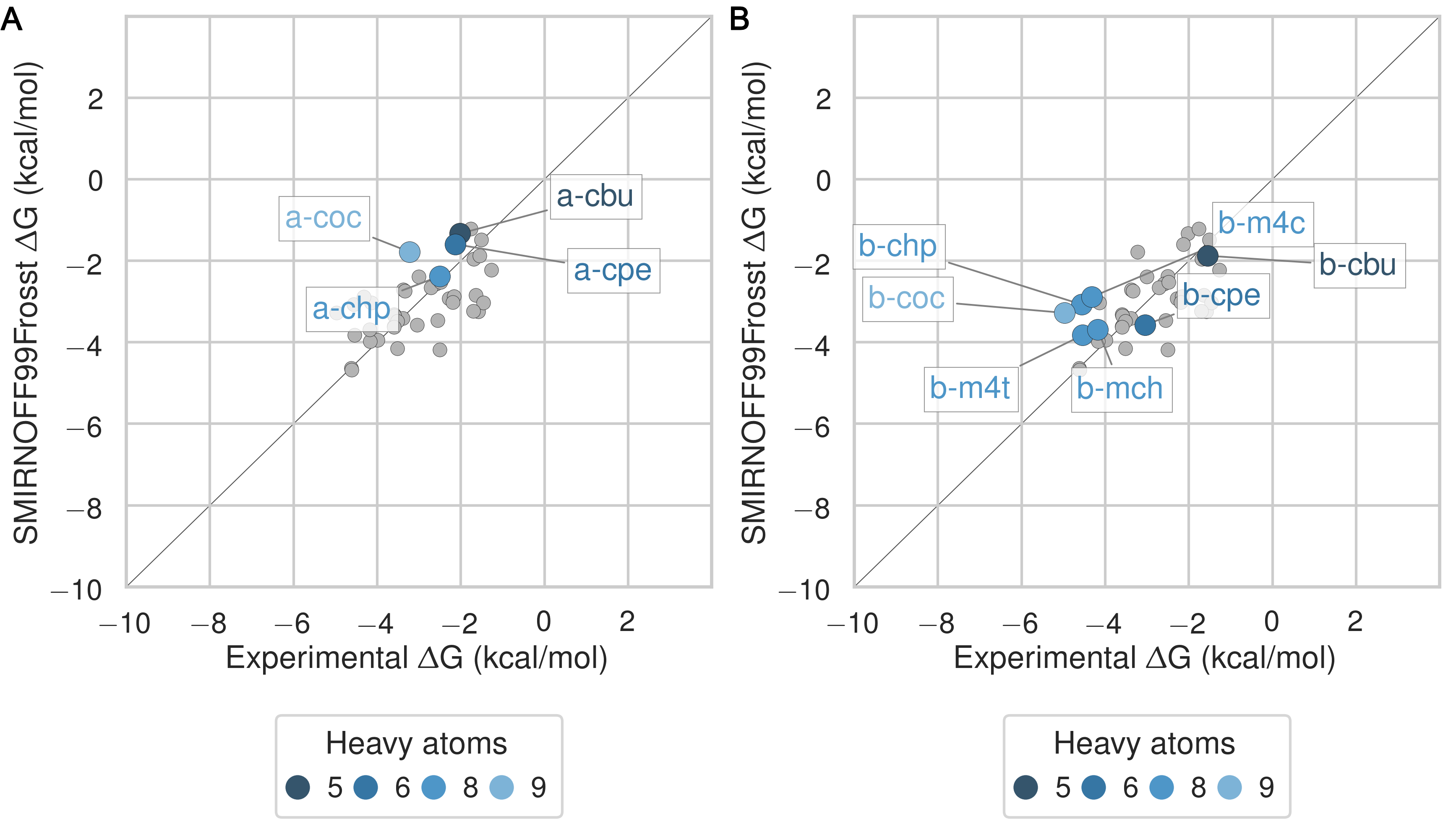
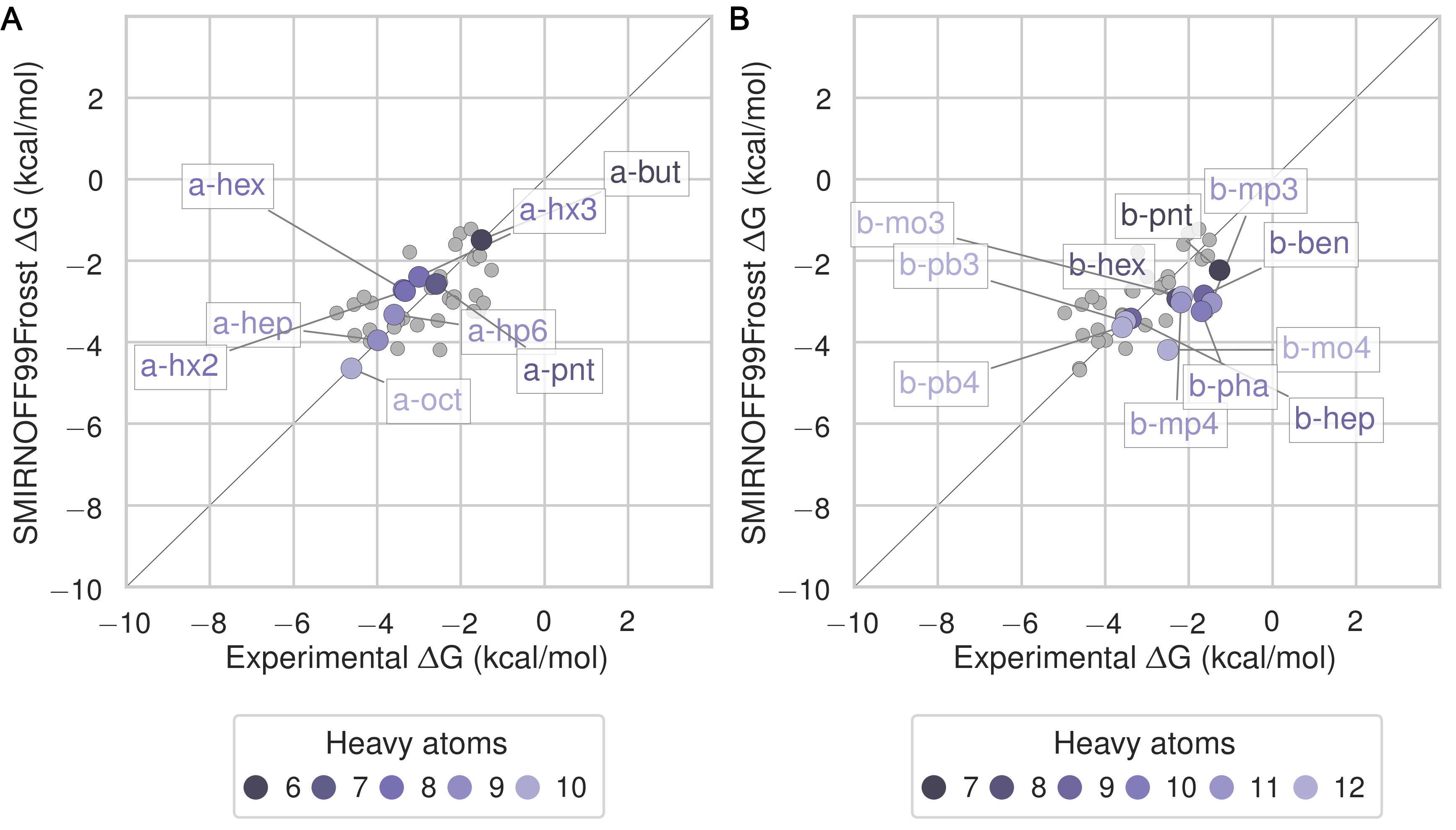
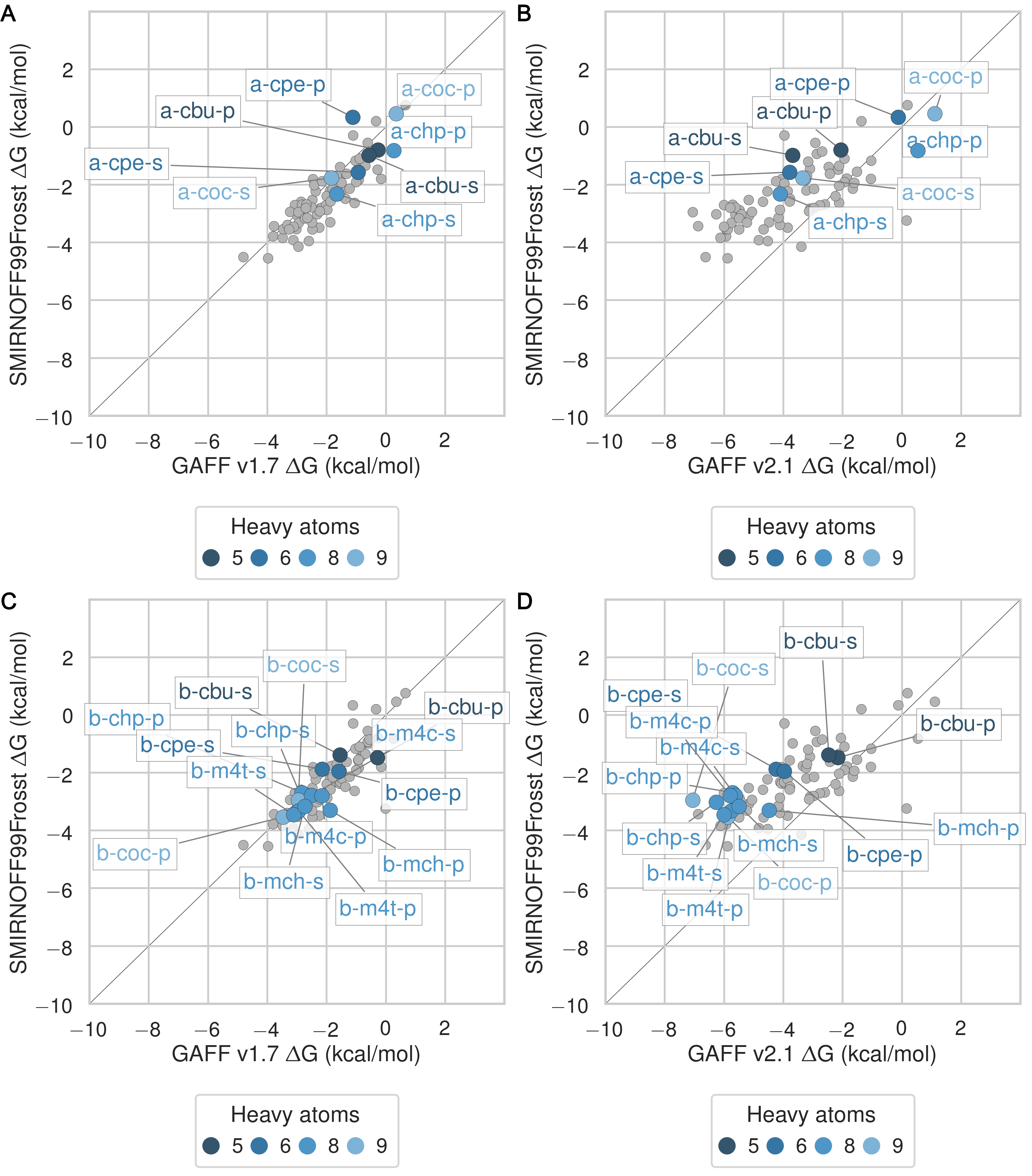
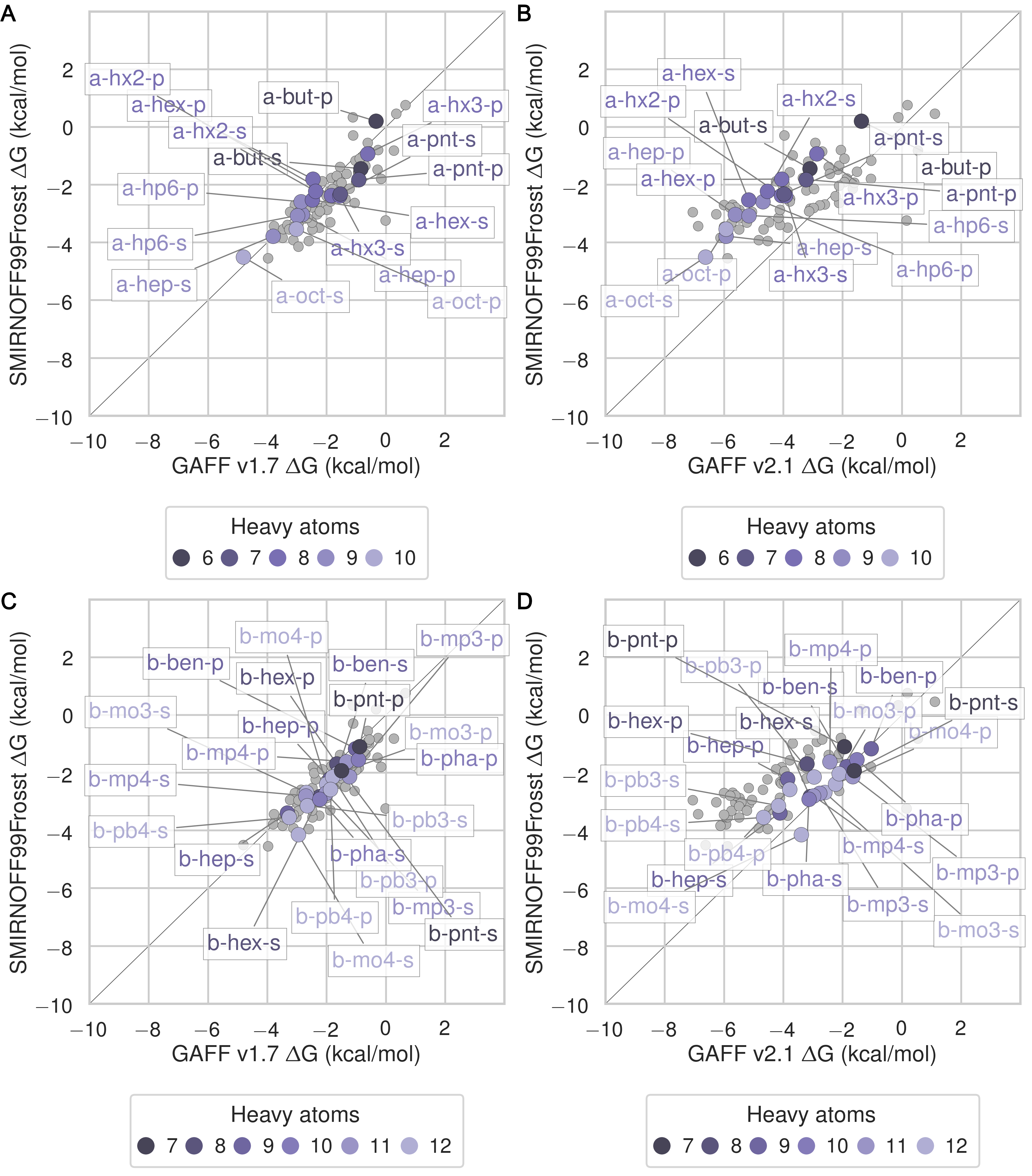
The SMIRNOFF99Frosst force field yields rather accurate binding free energies for binding of the ammonium guests (MSE = -0.1 [-0.5, 0.3] kcal/mol and RMSE = 0.7 [0.4, 1.1] kcal/mol) to both αCD and βCD (Figure 6 and Table 9). It also replicates the experimental trends that shorter-chain molecules bind less strongly, and that each guest binds more strongly to αCD than βCD. The results are also reasonably good for the cyclic alcohols (MSE = 0.7 [0.2, 1.2] kcal/mol and RMSE = 1.1 [0.7, 1.6] kcal/mol) (Figure 7 and Table 11), though the predicted affinities for αCD are uniformly too weak, while those for βCD are mostly too strong. Finally, SMIRNOFF99Frosst yields rather accurate binding affinities for the carboxylate guests with both αCD and βCD (MSE = -0.4 [-0.7, 0] kcal/mol and RMSE = 0.9 [0.6, 1.2] kcal/mmol) (Figure 8 and Table 10).
GAFF v1.7 tends to predict slightly weaker binding than SMIRNOFF99Frosst, whereas GAFF v2.1 predicts much stronger binding for all classses of guest compounds (Figures 25, 26, and 27).
Figure 6: Binding free energy (ΔG°) comparisons showing ammonium-containing guests in color and highlighted, for αCD (A) and βCD (B). Darker colors indicate shorter chain molecules. Non-ammonium guests are shown as smaller gray circles.Figure 7: Binding free energy (ΔG°) comparisons showing alcohol-containing guests in color and highlighted, for αCD (A) and βCD (B). Darker colors indicate smaller molecules. Non-alcohol guests are shown as smaller gray circles.Figure 8: Binding free energy (ΔG°) comparisons showing carboxylate-containing guests in color and highlighted, for αCD (A) and βCD (B). Darker colors indicates smaller molecules. Non-carboxylate guests are shown as smaller gray circles.
Differences in cyclodextrin force field parameters between SMIRNOFF99Frosst and GAFF
We now summarize differences among the parameters assigned to the host αCD by SMIRNOFF99Frosst, a descendant of parm99 and parm@Frosst; GAFF v1.7 (released circa March 2015 according to gaff.dat distributed with AMBER16); and GAFF v2.1 (which has not yet been published). On going from GAFF v1.7 to GAFF v2.1, the bond and angle parameters were updated to reproduce small molecule geometries obtained from high-level quantum mechanical calculations and vibrational spectra of over 600 molecules; the torsion parameters were optimized to reproduce the potential energy surfaces of torsion angles in 400 model compounds; and the Lennard-Jones coefficients were redeveloped to reproduce interaction energies and pure liquid properties, as specified in the footer of gaff2.dat provided with AmberTools18. Note that chemically analogous atoms, bonds, angles and torsions in αCD and βCD are assigned identical parameters.
Lennard-Jones
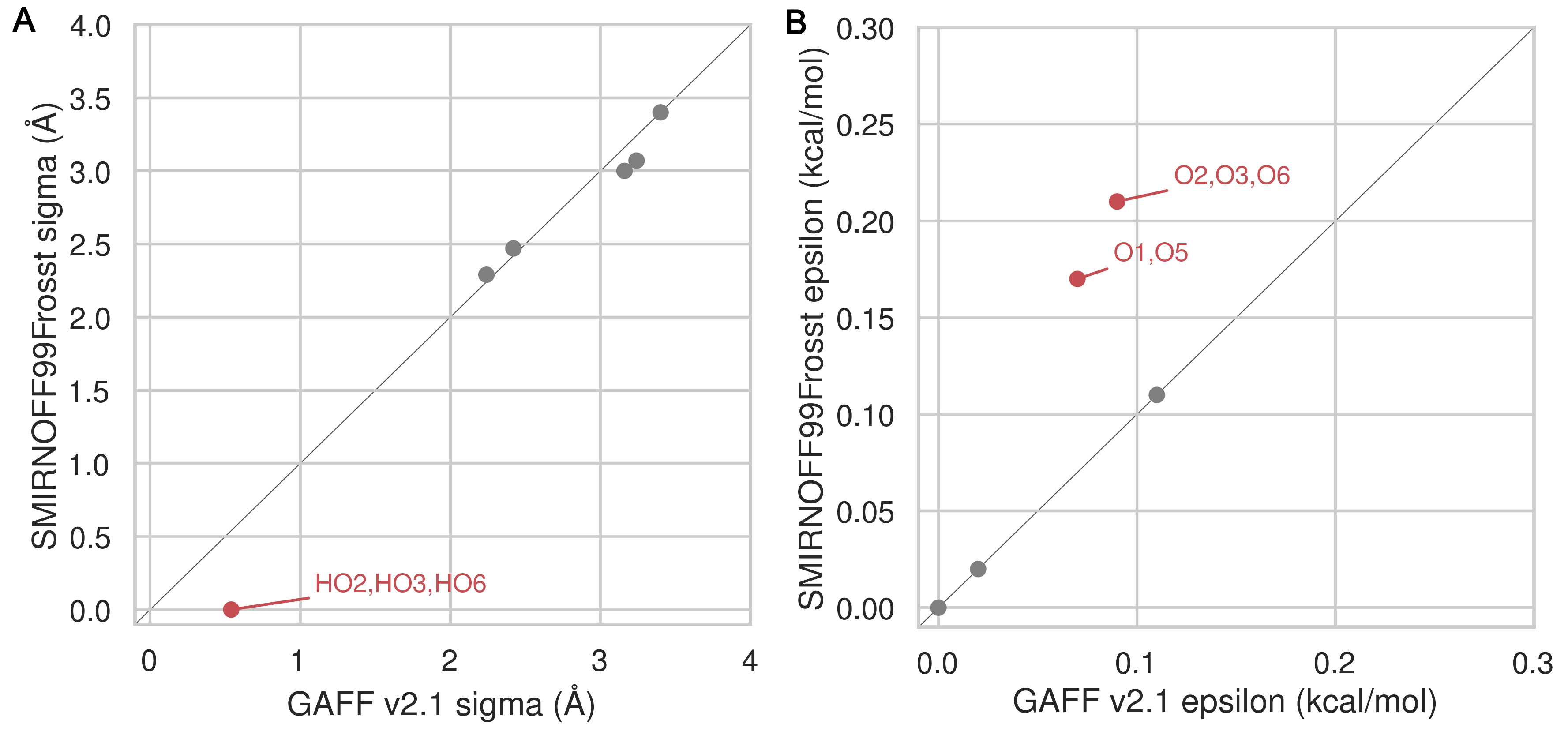
The SMIRNOFF99Frosst and GAFF v1.7 force fields assign identical σ and ε parameters to the atoms of αCD. Note, that hydroxyl hydrogens are assigned σ = 0 Å and ε = 0 kcal/mol in both GAFF v1.7 and SMIRNOFF99Frosst v1.0.5, but later versions of SMIRNOFF99Frosst, produced after the calculations in the current manuscript, adopt small σ and ε values based on a similiar atom type in parm@Frosst [48,49,50]. The GAFF v2.1 parameters differ in assigning shallower wells for oxygens and larger σ values for the hydroxyl hydrogens (Figure 9).
Figure 9: A comparison of Lennard-Jones nonbonded σ (A) and ε (B) parameters for SMIRNOFF99Frosst and GAFF v2.1. Values that differ by more than 10% are labeled in red. Atom names refer to Figure 2.
Bond stretches
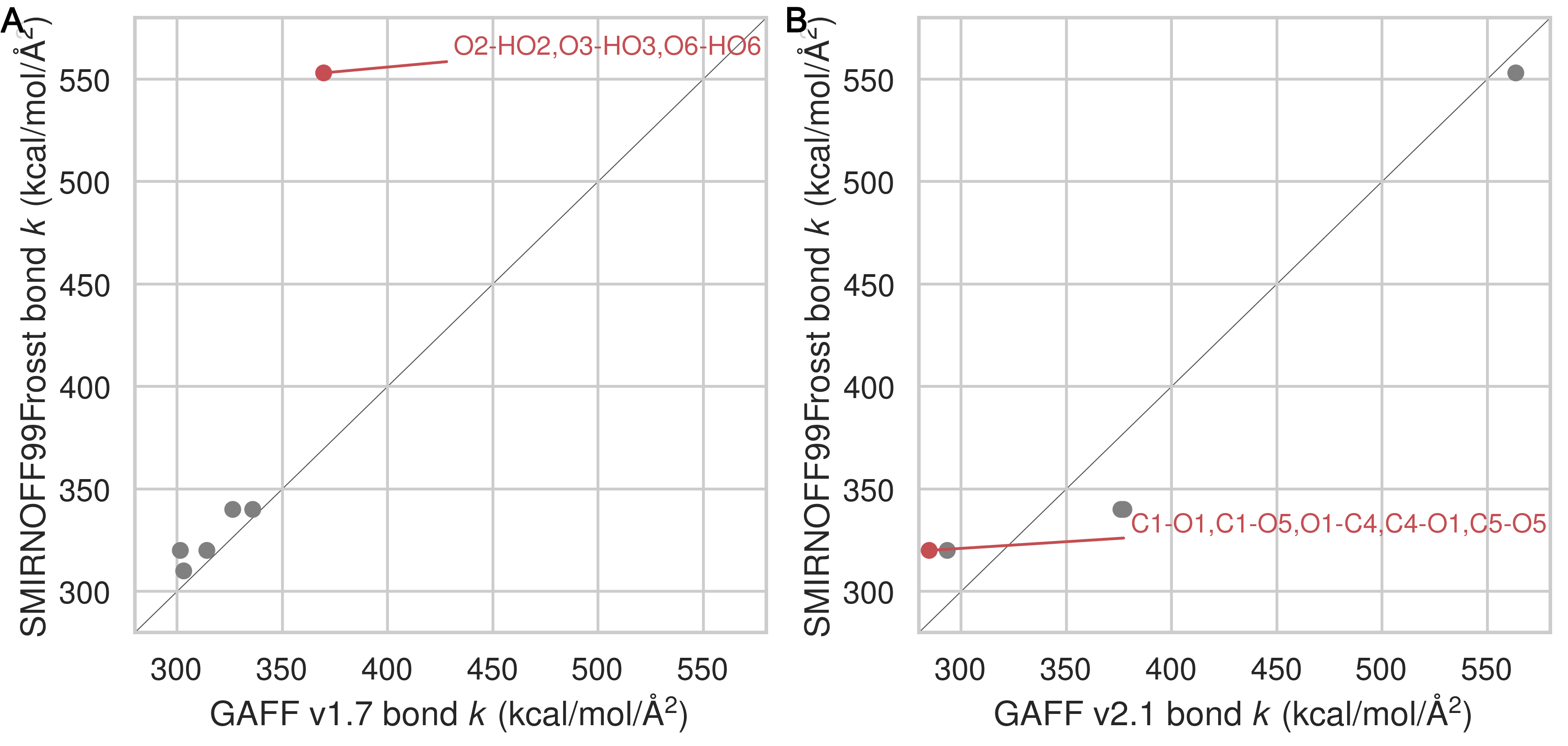
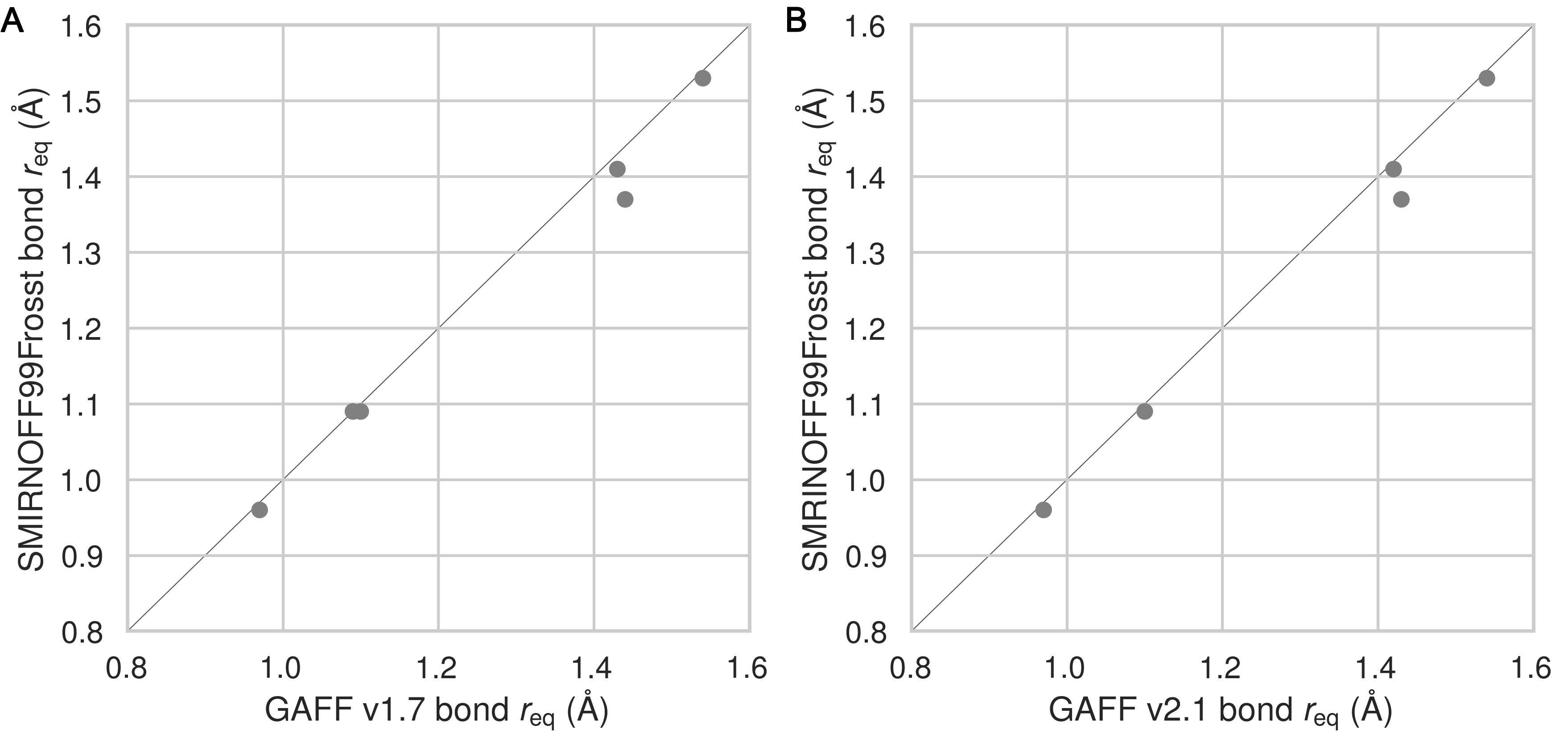
Equilibrium bond lengths are very similar among the three force fields (Figure 28), but there are noticeable differences among the force constants (Figure 10) Thus, compared to GAFF v1.7, SMIRNOFF99Frosst tends to have slightly larger bond force constants, except for the O–H hydroxyl bond force constant, which is much stronger. In GAFF v2.1, the O–H hydroxyl bond force constant is very close to that of SMIRNOFF99Frosst, but the carbon-oxygen bond constants are distinctly weaker.
Figure 10: A comparison of bond force constants between SMIRNOFF99Frosst and GAFF v1.7 (A), or SMIRNOFF99Frosst and GAFF v2.1 (B). Values that differ by more than 10% are labeled in red. Atom names refer to Figure 2.
Bond angles
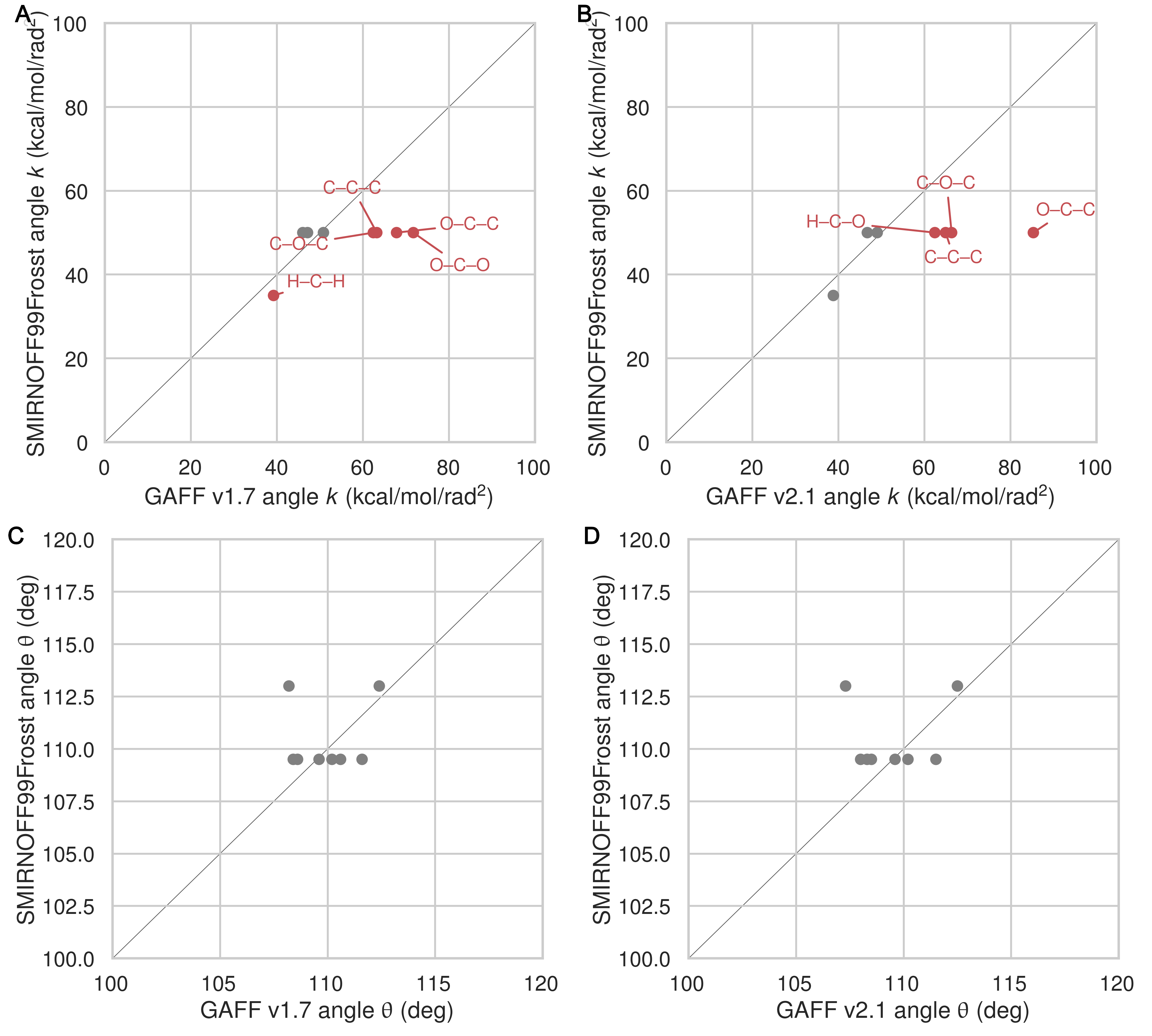
Relative to GAFF v1.7 and GAFF v2.1, SMIRNOFF99Frosst has fewer unique angle parameters applied to αCD; several distinct parameters appear to be compressed into a single force constant, around 50 kcal/mol/rad2 (Figure 11). These parameters correspond to C–C–C, C–O–C, O–C–O angles. The C–C–C angles are primarily around the ring of the glucose monomer. The C–O–C angles are both around the ring and between monomers (e.g., C1–O1–C4 and C1–O5–C5). Weaker force constants for these parameters in GAFF v1.7 compared to GAFF v2.1 may lead to increased flexibility.
Figure 11: A comparison of angle force constants between SMIRNOFF99Frosst and GAFF v1.7 (A) or SMIRNOFF99Frosst and GAFF v2.1 (B). A comparison of equilibrium angle values SMIRNOFF99Frosst and GAFF v1.7 (C) or SMIRNOFF99Frosst and GAFF v2.1 (D). Values that differ by more than 10% are labeled in red. Precise atom names have been omitted to compress multiple angles with the same parameter values into a single label.
Dihedral parameters
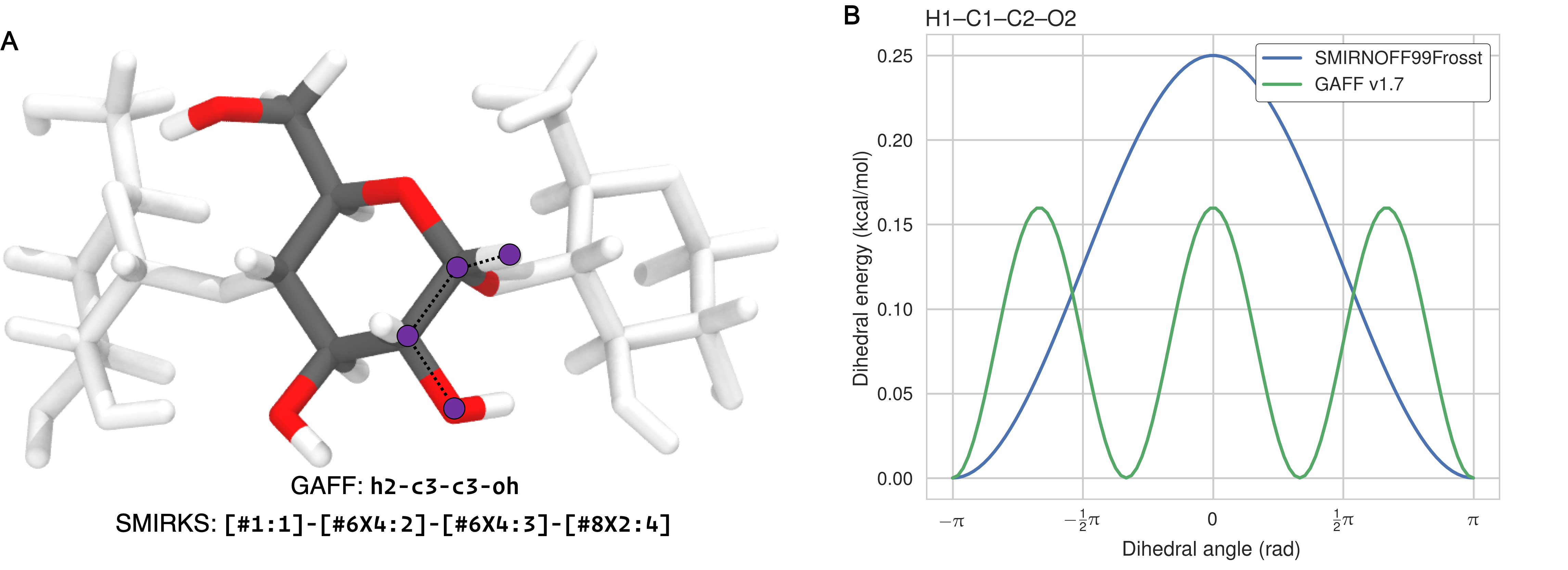
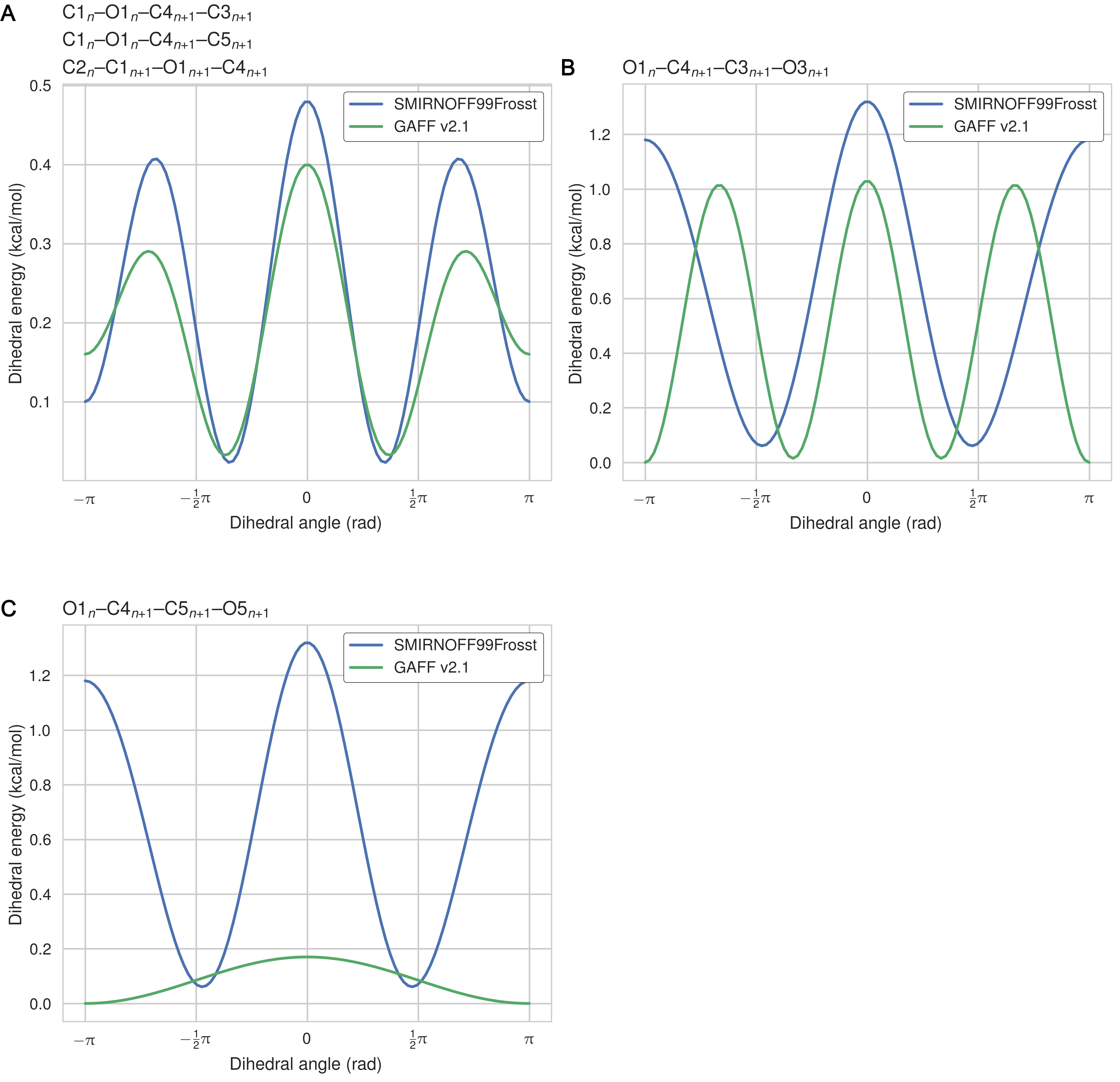
The dihedral parameters in SMIRNOFF99Frosst and GAFF v1.7 are extremely similar—where differences in barrier heights occur, they are in the hundredths or thousandths of 1 kcal/mol—with the exception of the H1–C1–C2–O2 parameter (Figure 2). For this dihedral, which corresponds to GAFF atom types h2-c3-c3-oh and SMIRKS pattern [#1:1]-[#6X4:2]-[#6X4:3]-[#8X2:4]), SMIRNOFF99Frosst applies a single term with periodicity = 1 and GAFF v1.7 applies a single term with periodicity = 3 (Table 12, Figures 12).
Figure 12: (A) The atoms in the H1–C1–C2–O2 dihedral marked in purple on a glucose monomer in cyclodextrin. (B) The dihedral energy term applied to H1–C1–C2–O2 in SMIRNOFF99Frosst and GAFF v1.7. Atom names refer to Figure 2.
The dihedral parameters in GAFF v2.1 differ from those in SMIRNOFF99Frosst in a number of ways. There are several dihedrals that have a different number of terms (Table 13). This is partly due to the addition of dihedral terms with a barrier height of exactly 0.00 kcal/mol in GAFF, which are used to override wildcard parameters that might match the same atom types. For example, GAFF v2.1 applies a three term energy function to the atom types c3-os-c3-c3, whereas SMIRNOFF99Frosst employs a two term energy function for the hydroxyl rotation SMIRKS pattern [#6X4:1]-[#6X4:2]-[#8X2H0:3]-[#6X4:4], but only the terms with periodicity 2 and 3 have nonzero barrier heights in GAFF v2.1. Similarly, SMIRNOFF99Frosst uses two nonzero terms to model the potential barrier for the SMIRKS pattern [#6X4:1]-[#6X4:2]-[#8X2H1:3]-[#1:4], yet GAFF v2.1 applies a single term with a barrier height of exactly 0.00 kcal/mol for this rotation (atom types c3-c3-oh-ho). The fact that GAFF employs dihedral terms with zero amplitude terms highlights the complexity that would be required to optimize existing force fields that have accumulated legacy parameters needed to maintain backwards compatibility with older force fields and simulation codes.
In other cases, SMIRNOFF99Frosst and GAFF v2.1 have disagreements on the barrier height after matching the periodicity and phase for a given dihedral. For example, the amplitudes for the O1–C1–O5–C5 dihedral are 1.35 kcal/mol and 0.97 kcal/mol for SMIRNOFF99Frosst and GAFF v2.1, respectively, for the term with periodicity = 1, whereas the amplitudes are 0.85 kcal/mol and 1.24 kcal/mol for SMIRNOFF99Frosst and GAFF v2.1, respectively, for the term with periodicity = 2. It is notable that the barrier heights in GAFF v2.1 are similiar in magnitude to those in SMIRNOFF99Frosst, yet GAFF v2.1 produces much more rigid structures (Table 3, Figure 14), as detailed in the following section. Moreoever, many of the dihedrals that act between a pair of neighboring glucose monomers (i.e., inter-residue dihedrals) in cyclodextrin differ in their periodicies, phases, and amplitudes between SMIRNOFF99Frosst and GAFF v2.1 (Table 4, Figure 13). The dihedral acting on atoms O1n–C4n+1–C5n+1–O5n+1 is quite significantly different, with multiple minima and and barrier heights. This dihedral partially controls the rotation of glucose monomers towards or away from the interior of the cyclodextrin cavity. Surprisingly, glucose monomers in GAFF v2.1 penetrate the open cavity much less frequently than in SMIRNOFF99Frosst, despite the lower and broader dihedral energy in GAFF v2.1.
Table 3: Dihedral barrier height differences between SMIRNOFF99Frosst and GAFF v2.1 for cases where the phase and periodicity of the energy term match but the barrier height does not. Atom names refer to Figure 2. Barrier height in kcal/mol.
SMIRNOFF99Frosst
GAFF v2.1
SMIRKS
Atom 1
Atom 2
Atom 3
Atom 4
Per
Phase
Height (kcal/mol)
Height (kcal/mol)
[#6X4:1]-[#6X4:2]-[#6X4:3]-[#6X4:4]
C1
C2
C3
C4
1
0
0.20
0.11
[#6X4:1]-[#6X4:2]-[#6X4:3]-[#6X4:4]
C1
C2
C3
C4
2
0
0.25
0.29
[#6X4:1]-[#6X4:2]-[#6X4:3]-[#6X4:4]
C1
C2
C3
C4
3
0
0.18
0.13
[*:1]-[#6X4:2]-[#6X4:3]-[*:4]
C1
C2
C3
O3
3
0
0.16
0.21
[*:1]-[#6X4:2]-[#8X2H0:3]-[*:4]
C1
O5
C5
H5
3
0
0.38
0.34
[#6X4:1]-[#6X4:2]-[#6X4:3]-[#6X4:4]
C2
C3
C4
C5
1
0
0.20
0.11
[#6X4:1]-[#6X4:2]-[#6X4:3]-[#6X4:4]
C2
C3
C4
C5
2
0
0.25
0.29
[#6X4:1]-[#6X4:2]-[#6X4:3]-[#6X4:4]
C2
C3
C4
C5
3
0
0.18
0.13
[#6X4:1]-[#6X4:2]-[#6X4:3]-[#6X4:4]
C3
C4
C5
C6
1
0
0.20
0.11
[#6X4:1]-[#6X4:2]-[#6X4:3]-[#6X4:4]
C3
C4
C5
C6
2
0
0.25
0.29
[#6X4:1]-[#6X4:2]-[#6X4:3]-[#6X4:4]
C3
C4
C5
C6
3
0
0.18
0.13
[*:1]-[#6X4:2]-[#6X4:3]-[*:4]
C4
C5
C6
O6
3
0
0.16
0.21
[#1:1]-[#6X4:2]-[#6X4:3]-[#1:4]
H1
C1
C2
H2
3
0
0.15
0.16
[#1:1]-[#6X4:2]-[#6X4:3]-[#1:4]
H2
C2
C3
H3
3
0
0.15
0.16
[*:1]-[#6X4:2]-[#8X2:3]-[#1:4]
H2
C2
O2
HO2
3
0
0.17
0.11
[#1:1]-[#6X4:2]-[#6X4:3]-[#1:4]
H3
C3
C4
H4
3
0
0.15
0.16
[*:1]-[#6X4:2]-[#8X2:3]-[#1:4]
H3
C3
O3
HO3
3
0
0.17
0.11
[#1:1]-[#6X4:2]-[#6X4:3]-[#1:4]
H4
C4
C5
H5
3
0
0.15
0.16
[#1:1]-[#6X4:2]-[#6X4:3]-[#1:4]
H5
C5
C6
H61
3
0
0.15
0.16
[#1:1]-[#6X4:2]-[#6X4:3]-[#1:4]
H5
C5
C6
H62
3
0
0.15
0.16
[#6X4:1]-[#8X2:2]-[#6X4:3]-[#8X2:4]
O1
C1
O5
C5
1
0
1.35
0.97
[#6X4:1]-[#8X2:2]-[#6X4:3]-[#8X2:4]
O1
C1
O5
C5
2
0
0.85
1.24
[#6X4:1]-[#8X2:2]-[#6X4:3]-[#8X2:4]
O1
C1
O5
C5
3
0
0.10
0.00
[*:1]-[#6X4:2]-[#6X4:3]-[*:4]
O2
C2
C3
C4
3
0
0.16
0.21
[#8X2:1]-[#6X4:2]-[#6X4:3]-[#8X2:4]
O2
C2
C3
O3
2
0
1.18
1.13
[#8X2:1]-[#6X4:2]-[#6X4:3]-[#8X2:4]
O2
C2
C3
O3
3
0
0.14
0.90
[*:1]-[#6X4:2]-[#6X4:3]-[*:4]
O3
C3
C4
C5
3
0
0.16
0.21
[*:1]-[#6X4:2]-[#8X2:3]-[#1:4]
H61
C6
O6
HO6
3
0
0.17
0.11
[*:1]-[#6X4:2]-[#8X2:3]-[#1:4]
H62
C6
O6
HO6
3
0
0.17
0.11
Table 4: Inter-residue dihedral parameter differences between SMIRNOFF99Frosst and GAFF v2.1. Atom names refer to Figure 2. NP: not present. Barrier height in kcal/mol.
SMIRNOFF99Frosst
GAFF v2.1
ID
Atom 1
Res 1
Atom 2
Res 2
Atom 3
Res 3
Atom 4
Res 4
Per
Phase
Height (kcal/mol)
Height (kcal/mol)
1
C1
n
O1
n
C4
n+1
C3
n+1
1
0
NP
0.00
C1
n
O1
n
C4
n+1
C3
n+1
2
0
0.10
0.16
C1
n
O1
n
C4
n+1
C3
n+1
3
0
0.38
0.24
2
C1
n
O1
n
C4
n+1
C5
n+1
1
0
NP
0.00
C1
n
O1
n
C4
n+1
C5
n+1
2
0
0.10
0.16
C1
n
O1
n
C4
n+1
C5
n+1
3
0
0.38
0.24
3
C2
n
C1
n+1
O1
n+1
C4
n+1
1
0
NP
0.00
C2
n
C1
n+1
O1
n+1
C4
n+1
2
0
0.10
0.16
C2
n
C1
n+1
O1
n+1
C4
n+1
3
0
0.38
0.24
4
O1
n
C4
n+1
C3
n+1
O3
n+1
1
0
NP
0.02
O1
n
C4
n+1
C3
n+1
O3
n+1
2
0
1.18
0.00
O1
n
C4
n+1
C3
n+1
O3
n+1
3
0
0.14
1.01
5
O1
n
C4
n+1
C5
n+1
O5
n+1
1
0
NP
0.17
O1
n
C4
n+1
C5
n+1
O5
n+1
2
0
1.18
0.00
O1
n
C4
n+1
C5
n+1
O5
n+1
3
0
0.14
0.00
Figure 13: The dihedral energy term applied to three inter-residue dihedrals in SMIRNOFF99Frosst and GAFF v2.1. Atom names refer to Figure 2.
There are no improper dihedrals in αCD or βCD, nor any of the guests.
Structural consequences of the force field parameter differences
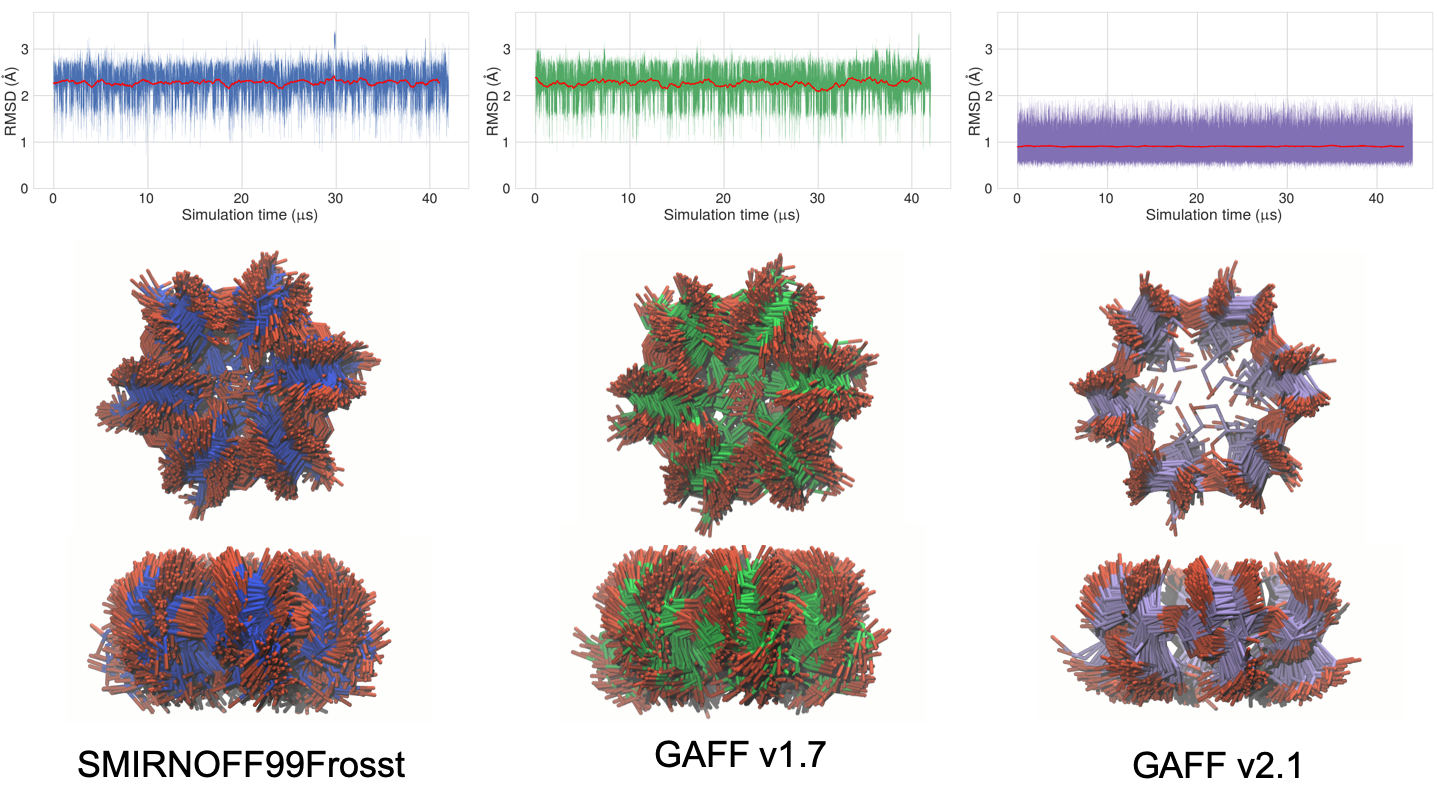
We observed a substantial difference between the conformational flexibility of the uncomplexed cyclodextrins in solution when simulated with GAFF v2.1 versus SMIRNOFF99Frosst and GAFF v1.7. With SMIRNOFF99Frosst and GAFF v1.7, the average RMSD of βCD, relative to the initial structure, is between 2.0–2.5 Å over 43 μs of unrestrained simulation, while with GAFF v2.1, the average RMSD is <1.0 Å (Figure 14). Not only are the RMSDs greater for SMIRNOFF99Frosst and GAFF v1.7, but there is greater variance in their RMSDs compared to GAFF v2.1, indicating greater flexibility. This large difference in structural fluctuations is clearly visible in the structure overlays also shown in the figure, which shows that GAFF v2.1 is the only one of the three force fields that leads to maintainance of a clearly defined binding cavity. In this respect, it is similar to the q4md-CD force field [51], which was designed specifically for cyclodextrins and which also maintains a relatively well-defined cavity [33].
Figure 14: Top: Root mean square deviation (RMSD in Å) of free βCD in the three force fields. Each RMSD is calculated relative to the initial structure, a gas-phase minimization of βCD with GAFF v1.7. A 1000 frame moving average is plotted in red. Middle: top-view of the unoccupied cavity of βCD with no guest (200 snapshots over 1 μs). Bottom: side-view of the unoccupied cavity. The carbons are colored blue in SMIRNOFF99Frosst, green in GAFF v1.7, and purple in GAFF v2.1. Hydrogen atoms have been hidden for clarity.
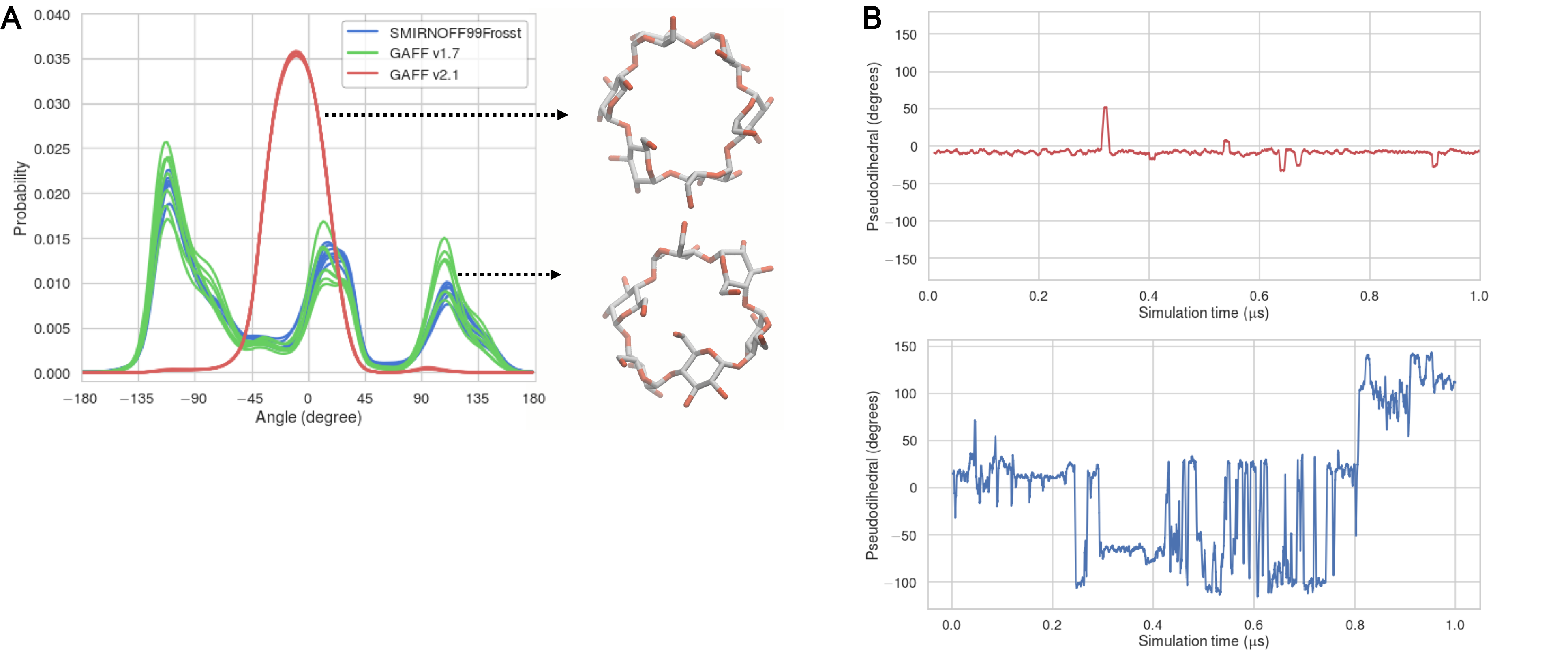
This difference may be further analyzed by considering the “flip” pseudodihedral O2n–C1n–C4n+1–O3n+1, which characterizes the orientation of glucose monomers relative to their neighbors. An angle of 0° corresponds approximately to a glucose that forms part of a cylindrical wall of the binding cavity, while an angle of ± 90° indicates a glucose that has flipped to put its plane parallel to the top and bottom of the cylinder, partly filling the cavity. This dihedral is tightly distributed in GAFF v2.1, with all seven instances having a Gaussian-like distribution centered around -10° (Figure 15, A). GAFF v1.7 and SMIRNOFF99Frosst display, a mixed population of monomers both aligned with, and perpendicular to, the cyclodextrin cavity. In particular, during a single 1 μs simulation, each monomer will sample conformations at 0° and ±90°, as indicated by the timeseries in Figure 15, B. As detailed in the Discussion, the less flexible representation afforded by GAFF v2.1 agrees better with available NMR and crystallographic data.
Figure 15: (A) Population histograms of the pseudodihedral in free βCD, averaged over 43 μs, for each force field; one curve is drawn for each pseudodihedral in βCD. Representative structures for SMIRNOFF99Frosst and GAFF v2.1 are indicated by the black arrows. (B) Timeseries of a psuedodihedral in a GAFF v2.1 simulation (red), or SMIRNOFF99Frosst simulation (blue).
Discussion
As a terse representation of a GAFF-like force field, SMIRNOFF99Frosst performs remarkably well. Despite having far fewer parameters than GAFF v1.7 and GAFF v2.1, SMIRNOFF99Frosst performs as well as GAFF v1.7 and arguably better than GAFF v2.1 on estimated binding free energies of small molecules to αCD and βCD, based on the mean signed error relative to experiment. Moreover, SMIRNOFF99Frosst performs better than either GAFF v1.7 or GAFF v2.1 on predicted binding enthalpies, with a mean signed error less than 1 kcal/mol. It should be noted that the binding free energy and enthalpy root mean squared errors (RMSE) and mean signed errors (MSE) for GAFF v2.1 are not substantially worse than those of SMIRNOFF99Frosst, and GAFF v2.1 has statistically significant better correlations with the the experimental data. GAFF v2.1 has excellent agreement with experiment on predicted binding entropy, followed by SMIRNOFF99Frosst and then GAFF v1.7. Taken together, these results support the notion that a force field with many fewer parameters can provide competitive performance. The reduction in the number of parameters, and the simplification of the force field specification, will make it easier to iteratively refine and optimize SMIRNOFF99Frosst against experimental data and the results of quantum mechanical calculations.
However, both SMIRNOFF99Frosst and GAFF v1.7 result in excessively flexible representations of the cyclodextrin hosts, as detailed below. Cézard, et al. present strong NMR evidence that the vicinal 3J H5–H6′ (atom names H5–H62 in Figure 2) and 3J H5-H6″(atom names H5–H61) coupling show minimal fluctuation in distance over a number of timescales, suggesting little change in the population of rotamers [51]. This is also evident in X-ray structures, where the rigidity of the cyclodextrin ring is retained as long as water is present in the cavity and the torsional angles between adjacent glucose units show little variance (0.3–0.6°) across different crystal structures [52]. The combination of X-ray and NMR data suggest that the specialized q4md-CD [51] force field, and the rigid GAFF v2.1 [33] force field, better model the flexibility of the CD cavity. The CHARMM36 force field displays similar structural dynamics to q4md-CD, with certain GROMOS force fields even more rigid than those [53]. The present results suggest that, as SMIRNOFF99Frosst is further developed, it will be important to include sugars and other carbohydrates in the training sets used to develop parameters. Unfortunately, it may be challenging to find the types of high quality experimental data typically used to train force fields—heats of vaporization, heats of mixing, hydration free energies, and partition coefficients, among others—for biologically relevant sugars. Proper accounting of sugars, and protein-sugar interactions, will be especially useful for modeling physiologically relevant protein structures, such as proteoglycans and glycopeptides.
The greater rigidity of the cyclodextrins when simulated with GAFF v2.1 may contribute to its tendency to generate greater binding affinities and more negative enthalpies than the other two force fields, as a more rigid host may avoid an energy penalty associated with flipping the glucose residues out of the binding cavity to accommodate a guest molecule. The better preorganized cavity might also relate to the uniformly higher correlations between calculation and experiment for GAFF v2.1. On the other hand, it is perhaps unexpected that this force field which best represents the conformational preferences of the cyclodextrin yields consistently too negative binding free energies and enthalpies. It is worth noting the magnitude of these effects will depend on the guest parameters, as well as water model and ion parameters as well.
More broadly, the results presented in this manuscript further demonstrate that host-guest binding thermodynamics can be used to benchmark force fields, to help diagnose issues with parameters applied to specific functional groups, and to suggest directions for improvements. We are therefore continuing to build out experimental host-guest datasets tuned for this purpose, and to further streamline host-guest binding thermodynamics calculations so that binding data can be used alongside other data types, such as liquid properties, by automated tools for optimizing force field parameters.
Code and data availability
GitHub repository used to convert AMBER input files from GAFF force field to SMIRNOFF99Frosst.
GitHub repository for setting up the attach-pull-release calculations using paprika version 0.0.3.
GitHub repository for analyzing the simulations and generating the plots in this manuscript.
GitHub repository for the Open Force Field group containing the toolkit and force field XML file.
This work was funded in part by grant GM061300 to MKG from the National Institute of General Medical Sciences of the NIH. JDC was funded in part by grants R01 GM121505 and R01 GM124270 from the National Institute of General Medical Sciences of the NIH and P30 CA008748 from the National Cancer Institute of the NIH. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Disclosures
The authors declare the following competing financial interest(s): MKG has an equity interest in and is a cofounder and scientific advisor of VeraChem LLC. JDC is a member of the Scientific Advisory Board of OpenEye Scientific Software. The Chodera laboratory receives or has received funding from multiple sources, including the National Institutes of Health, the National Science Foundation, the Parker Institute for Cancer Immunotherapy, Relay Therapeutics, Entasis Therapeutics, Silicon Therapeutics, EMD Serono (Merck KGaA), AstraZeneca, XtalPi, the Molecular Sciences Software Institute, the Starr Cancer Consortium, the Open Force Field Consortium, Cycle for Survival, a Louis V. Gerstner Young Investigator Award, the Einstein Foundation, and the Sloan Kettering Institute. A complete funding history for the Chodera lab can be found at http://choderalab.org/funding.
List of abbreviations
APR, attach-pull-release; CD, cyclodextrin; GAFF, Generalized AMBER Force Field
References
1. Overview of the SAMPL6 host–guest binding affinity prediction challenge
Andrea Rizzi, Steven Murkli, John N. McNeill, Wei Yao, Matthew Sullivan, Michael K. Gilson, Michael W. Chiu, Lyle Isaacs, Bruce C. Gibb, David L. Mobley, John D. Chodera Journal of Computer-Aided Molecular Design (2018-10) https://doi.org/gfpzh5
DOI: 10.1007/s10822-018-0170-6 · PMID: 30415285 · PMCID: PMC6301044
2. Attach-Pull-Release Calculations of Ligand Binding and Conformational Changes on the First BRD4 Bromodomain
Germano Heinzelmann, Niel M. Henriksen, Michael K. Gilson Journal of Chemical Theory and Computation (2017-05-31) https://doi.org/gbpj4m
DOI: 10.1021/acs.jctc.7b00275 · PMID: 28564537 · PMCID: PMC5541932
3. Computation of protein–ligand binding free energies using quantum mechanical bespoke force fields
Daniel J. Cole, Israel Cabeza de Vaca, William L. Jorgensen MedChemComm (2019) https://doi.org/gfz353
DOI: 10.1039/c9md00017h · PMID: 31391883 · PMCID: PMC6644397
4. Predictions of Ligand Selectivity from Absolute Binding Free Energy Calculations
Matteo Aldeghi, Alexander Heifetz, Michael J. Bodkin, Stefan Knapp, Philip C. Biggin Journal of the American Chemical Society (2017-01-09) https://doi.org/f9kr6h
DOI: 10.1021/jacs.6b11467 · PMID: 28009512 · PMCID: PMC5253712
5. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field
Lingle Wang, Yujie Wu, Yuqing Deng, Byungchan Kim, Levi Pierce, Goran Krilov, Dmitry Lupyan, Shaughnessy Robinson, Markus K. Dahlgren, Jeremy Greenwood, … Robert Abel Journal of the American Chemical Society (2015-02-12) https://doi.org/f64pdz
DOI: 10.1021/ja512751q · PMID: 25625324
6. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules
Katarina Roos, Chuanjie Wu, Wolfgang Damm, Mark Reboul, James M. Stevenson, Chao Lu, Markus K. Dahlgren, Sayan Mondal, Wei Chen, Lingle Wang, … Edward D. Harder Journal of Chemical Theory and Computation (2019-02-15) https://doi.org/gfvpnf
DOI: 10.1021/acs.jctc.8b01026 · PMID: 30768902
7. Validation of AMBER/GAFF for Relative Free Energy Calculations
Lin Song, Tai-Sung Lee, Chun Zhu, Darrin M. York, Kenneth M. Merz Jr. American Chemical Society (ACS) (2019-02-04) https://doi.org/gf22kq
DOI: 10.26434/chemrxiv.7653434
8. Overview of the SAMPL5 host–guest challenge: Are we doing better?
Jian Yin, Niel M. Henriksen, David R. Slochower, Michael R. Shirts, Michael W. Chiu, David L. Mobley, Michael K. Gilson Journal of Computer-Aided Molecular Design (2016-09-22) https://doi.org/f9m82x
DOI: 10.1007/s10822-016-9974-4 · PMID: 27658802 · PMCID: PMC5241188
9. The Movable Type Method Applied to Protein–Ligand Binding
Zheng Zheng, Melek N. Ucisik, Kenneth M. Merz Journal of Chemical Theory and Computation (2013-11-26) https://doi.org/f5kjt2
DOI: 10.1021/ct4005992 · PMID: 24535920 · PMCID: PMC3924725
10. Blinded predictions of standard binding free energies: lessons learned from the SAMPL6 challenge
Michail Papadourakis, Stefano Bosisio, Julien Michel Journal of Computer-Aided Molecular Design (2018-08-29) https://doi.org/gfpbrc
DOI: 10.1007/s10822-018-0154-6 · PMID: 30159717
11. Resolving the problem of trapped water in binding cavities: prediction of host–guest binding free energies in the SAMPL5 challenge by funnel metadynamics
Soumendranath Bhakat, Pär Söderhjelm Journal of Computer-Aided Molecular Design (2016-08-29) https://doi.org/f9m894
DOI: 10.1007/s10822-016-9948-6 · PMID: 27573983 · PMCID: PMC5239820
12. Absolute binding free energies for octa-acids and guests in SAMPL5
Florentina Tofoleanu, Juyong Lee, Frank C. Pickard IV, Gerhard König, Jing Huang, Minkyung Baek, Chaok Seok, Bernard R. Brooks Journal of Computer-Aided Molecular Design (2016-09-30) https://doi.org/f9m9c2
DOI: 10.1007/s10822-016-9965-5 · PMID: 27696242 · PMCID: PMC6472255
13. Computational Calorimetry: High-Precision Calculation of Host–Guest Binding Thermodynamics
Niel M. Henriksen, Andrew T. Fenley, Michael K. Gilson Journal of Chemical Theory and Computation (2015-08-26) https://doi.org/f7q3mj
DOI: 10.1021/acs.jctc.5b00405 · PMID: 26523125 · PMCID: PMC4614838
14. Prediction of CB[8] host–guest binding free energies in SAMPL6 using the double-decoupling method
Kyungreem Han, Phillip S. Hudson, Michael R. Jones, Naohiro Nishikawa, Florentina Tofoleanu, Bernard R. Brooks Journal of Computer-Aided Molecular Design (2018-08-06) https://doi.org/gfpzp2
DOI: 10.1007/s10822-018-0144-8 · PMID: 30084077 · PMCID: PMC6347468
15. Comparison of the umbrella sampling and the double decoupling method in binding free energy predictions for SAMPL6 octa-acid host–guest challenges
Naohiro Nishikawa, Kyungreem Han, Xiongwu Wu, Florentina Tofoleanu, Bernard R. Brooks Journal of Computer-Aided Molecular Design (2018-10) https://doi.org/gfz352
DOI: 10.1007/s10822-018-0166-2 · PMID: 30324304 · PMCID: PMC6413509
16. Detailed potential of mean force studies on host–guest systems from the SAMPL6 challenge
Lin Frank Song, Nupur Bansal, Zheng Zheng, Kenneth M. Merz Journal of Computer-Aided Molecular Design (2018-08-24) https://doi.org/gfpw5t
DOI: 10.1007/s10822-018-0153-7 · PMID: 30143917
17. The SAMPL4 host–guest blind prediction challenge: an overview
Hari S. Muddana, Andrew T. Fenley, David L. Mobley, Michael K. Gilson Journal of Computer-Aided Molecular Design (2014-03-06) https://doi.org/f5585s
DOI: 10.1007/s10822-014-9735-1 · PMID: 24599514 · PMCID: PMC4053502
18. The statistical-thermodynamic basis for computation of binding affinities: a critical review
M. K. Gilson, J. A. Given, B. L. Bush, J. A. McCammon Biophysical Journal (1997-03) https://doi.org/dkv3sk
DOI: 10.1016/s0006-3495(97)78756-3 · PMID: 9138555 · PMCID: PMC1184492
19. Bridging Calorimetry and Simulation through Precise Calculations of Cucurbituril–Guest Binding Enthalpies
Andrew T. Fenley, Niel M. Henriksen, Hari S. Muddana, Michael K. Gilson Journal of Chemical Theory and Computation (2014-08) https://doi.org/f6hv4x
DOI: 10.1021/ct5004109 · PMID: 25221445 · PMCID: PMC4159218
20. Structures of the Common Cyclodextrins and Their Larger AnaloguesBeyond the Doughnut
Wolfram Saenger, Joël Jacob, Katrin Gessler, Thomas Steiner, Daniel Hoffmann, Haruyo Sanbe, Kyoko Koizumi, Steven M. Smith, Takeshi Takaha Chemical Reviews (1998-07) https://doi.org/dgz7hm
DOI: 10.1021/cr9700181
21. Toward Expanded Diversity of Host–Guest Interactions via Synthesis and Characterization of Cyclodextrin Derivatives
K. Kellett, S. A. Kantonen, B. M. Duggan, M. K. Gilson Journal of Solution Chemistry (2018-06-04) https://doi.org/gfp949
DOI: 10.1007/s10953-018-0769-1
23. Escaping Atom Types in Force Fields Using Direct Chemical Perception
David L. Mobley, Caitlin C. Bannan, Andrea Rizzi, Christopher I. Bayly, John D. Chodera, Victoria T. Lim, Nathan M. Lim, Kyle A. Beauchamp, David R. Slochower, Michael R. Shirts, … Peter K. Eastman Journal of Chemical Theory and Computation (2018-10-11) https://doi.org/gffnf3
DOI: 10.1021/acs.jctc.8b00640 · PMID: 30351006 · PMCID: PMC6245550
25. A Modified Version of the Cornellet al.Force Field with Improved Sugar Pucker Phases and Helical Repeat
Thomas E. Cheatham III, Piotr Cieplak, Peter A. Kollman Journal of Biomolecular Structure and Dynamics (1999-02) https://doi.org/gfzp4j
DOI: 10.1080/07391102.1999.10508297 · PMID: 10217454
27. AMBER 2018
D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. III Cheatham, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilson, … P. A. Kollman
29. Development and testing of a general amber force field
Junmei Wang, Romain M. Wolf, James W. Caldwell, Peter A. Kollman, David A. Case Journal of Computational Chemistry (2004) https://doi.org/cdmcnb
DOI: 10.1002/jcc.20035 · PMID: 15116359
30. A general small molecule force field descended from AMBER99 and parm@Frosst, available in the SMIRNOFF format: openforcefield/smirnoff99Frosst
Open Force Field Initiative
(2019-09-27) https://github.com/openforcefield/smirnoff99Frosst
32. Thermodynamic and Nuclear Magnetic Resonance Study of the Reactions of α- and β-Cyclodextrin with Acids, Aliphatic Amines, and Cyclic Alcohols
Mikhail V. Rekharsky, Martin P. Mayhew, Robert N. Goldberg, Philip D. Ross, Yuko Yamashoji, Yoshihisa Inoue The Journal of Physical Chemistry B (1997-01) https://doi.org/cmwfwn
DOI: 10.1021/jp962715n
33. Evaluating Force Field Performance in Thermodynamic Calculations of Cyclodextrin Host–Guest Binding: Water Models, Partial Charges, and Host Force Field Parameters
Niel M. Henriksen, Michael K. Gilson Journal of Chemical Theory and Computation (2017-07-11) https://doi.org/gd2z2t
DOI: 10.1021/acs.jctc.7b00359 · PMID: 28696692 · PMCID: PMC5606194
34. Chiral Recognition Thermodynamics of β-Cyclodextrin: The Thermodynamic Origin of Enantioselectivity and the Enthalpy−Entropy Compensation Effect
Mikhail Rekharsky, Yoshihisa Inoue Journal of the American Chemical Society (2000-05) https://doi.org/c2tvdg
DOI: 10.1021/ja9921118
36. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation
Araz Jakalian, David B. Jack, Christopher I. Bayly Journal of Computational Chemistry (2002-10-18) https://doi.org/cktk6g
DOI: 10.1002/jcc.10128 · PMID: 12395429
38. Comparison of simple potential functions for simulating liquid water
William L. Jorgensen, Jayaraman Chandrasekhar, Jeffry D. Madura, Roger W. Impey, Michael L. Klein The Journal of Chemical Physics (1983-07-15) https://doi.org/dg9sq8
DOI: 10.1063/1.445869
39. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations
In Suk Joung, Thomas E. Cheatham III The Journal of Physical Chemistry B (2008-07) https://doi.org/cgwnj7
DOI: 10.1021/jp8001614 · PMID: 18593145 · PMCID: PMC2652252
40. Lessons learned from comparing molecular dynamics engines on the SAMPL5 dataset
Michael R. Shirts, Christoph Klein, Jason M. Swails, Jian Yin, Michael K. Gilson, David L. Mobley, David A. Case, Ellen D. Zhong Journal of Computer-Aided Molecular Design (2016-10-27) https://doi.org/f9m3wn
DOI: 10.1007/s10822-016-9977-1 · PMID: 27787702 · PMCID: PMC5581938
41. Overcoming dissipation in the calculation of standard binding free energies by ligand extraction
Camilo Velez-Vega, Michael K. Gilson Journal of Computational Chemistry (2013-08) https://doi.org/gbdv9w
DOI: 10.1002/jcc.23398 · PMID: 24038118 · PMCID: PMC3932244
43. Statistically optimal analysis of samples from multiple equilibrium states
Michael R. Shirts, John D. Chodera The Journal of Chemical Physics (2008-09-28) https://doi.org/cvzgk7
DOI: 10.1063/1.2978177 · PMID: 19045004 · PMCID: PMC2671659
45. Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning
Chad W. Hopkins, Scott Le Grand, Ross C. Walker, Adrian E. Roitberg Journal of Chemical Theory and Computation (2015-03-30) https://doi.org/f697mk
DOI: 10.1021/ct5010406 · PMID: 26574392
46. Nonlinear scaling schemes for Lennard-Jones interactions in free energy calculations
Thomas Steinbrecher, David L. Mobley, David A. Case The Journal of Chemical Physics (2007-12-07) https://doi.org/fc7n55
DOI: 10.1063/1.2799191 · PMID: 18067350
47. Error estimates on averages of correlated data
H. Flyvbjerg, H. G. Petersen The Journal of Chemical Physics (1989-07) https://doi.org/bjpm5j
DOI: 10.1063/1.457480
51. Molecular dynamics studies of native and substituted cyclodextrins in different media: 1. Charge derivation and force field performances
Christine Cézard, Xavier Trivelli, Frédéric Aubry, Florence Djedaïni-Pilard, François-Yves Dupradeau Physical Chemistry Chemical Physics (2011) https://doi.org/cv3hss
DOI: 10.1039/c1cp20854c · PMID: 21792425
52. Topography of cyclodextrin inclusion complexes. 8. Crystal and molecular structure of the .alpha.-cyclodextrin-methanol-pentahydrate complex. Disorder in a hydrophobic cage
B. Hingerty, W. Saenger Journal of the American Chemical Society (1976-05) https://doi.org/cmfz5v
DOI: 10.1021/ja00427a050
53. Validation and Comparison of Force Fields for Native Cyclodextrins in Aqueous Solution
Julia Gebhardt, Catharina Kleist, Sven Jakobtorweihen, Niels Hansen The Journal of Physical Chemistry B (2018-01-24) https://doi.org/gcv3xq
DOI: 10.1021/acs.jpcb.7b11808 · PMID: 29287148
Supporting Information
Figure 16: Comparison of AM1-BCC partial atomic charges assigned by running antechamber on a single glucose monomer (A) or on an entire αCD molecule (B) with the option -pl 10 to specify the maximum path length used to determine the equivalence of atomic charges.Figure 17: Comparison of calculated absolute binding entropies (−TΔS°) with experiment with SMIRNOFF99Frosst parameters (top), GAFF v1.7 parameters (middle), or GAFF v2.1 parameters (bottom) applied to both host and guest. The orange, blue, and purple coloring distinguish the functional group of the guest as an ammonium, alcohol, or carboxylate, respectively.Figure 18: The primary (left) and secondary (right) face of αCD.Figure 19: Binding free energies (ΔG°) with the primary orientation results colored in blue and secondary orientation results colored in green.Figure 20: Binding free energies (ΔG°) replotted from Figure 19 with points whose difference in binding free energy along either axis is greater than 2 kcal/mol shown in color. Arrows point from ΔG° for the secondary to ΔG° for the primary cavity.Figure 21: Comparison of calculated absolute binding free energies (ΔG°) between force field combinations. The orange, blue, and purple coloring distinguish the functional group of the guest as an ammonium, alcohol, or carboxylate, respectively.Figure 22: Comparison of calculated absolute binding free enthalpies (ΔH) between force field combinations. The orange, blue, and purple coloring distinguish the functional group of the guest as an ammonium, alcohol, or carboxylate, respectively.Figure 23: Comparison of calculated absolute binding entropies (−TΔS°) between force field combinations. The orange, blue, and purple coloring distinguish the functional group of the guest as an ammonium, alcohol, or carboxylate, respectively.Figure 24: Binding free energies (ΔG°) replotted from Figure 3, with αCD points colored in blue and βCD points in grey (left) or αCD points in grey with βCD points colored in green (right).Figure 25: Binding free energy (ΔG°) comparisons showing ammonium guests in color and highlighted.Figure 26: Binding free energy (ΔG°) comparisons showing alcohols guests in color and highlighted.Figure 27: Binding free energy (ΔG°) comparisons showing carboxylates guests in color and highlighted.Figure 28: A comparison of bond equilibrium lengths for SMIRNOFF99Frosst, GAFF v1.7, and GAFF v2.1. Atom names refer to Figure 2.
Table 5: Experimental and predicted binding free energies (ΔG°). Values in kcal/mol.
System
Experimental
SMIRNOFF99Frosst
GAFF v1.7
GAFF v2.1
Mean
SEM
Mean
SEM
Mean
SEM
Mean
SEM
a-bam
-1.58
0.02
-3.25
0.44
-0.82
0.21
-2.93
0.23
a-but
-1.51
0.04
-1.49
0.27
-1.09
0.20
-3.14
0.22
a-cbu
-2.02
0.02
-1.33
0.19
-0.89
0.22
-3.73
0.21
a-chp
-2.51
0.06
-2.38
0.28
-1.69
0.24
-4.11
0.23
a-coc
-3.23
1.14
-1.78
0.29
-1.86
0.24
-3.35
0.24
a-cpe
-2.13
0.02
-1.59
0.25
-1.50
0.29
-3.79
0.22
a-ham
-3.53
0.00
-3.43
0.30
-3.02
0.19
-5.99
0.17
a-hep
-3.99
0.01
-3.95
0.21
-3.93
0.20
-6.23
0.17
a-hex
-3.38
0.01
-2.70
0.21
-2.92
0.21
-5.27
0.20
a-hp6
-3.60
0.00
-3.32
0.23
-3.37
0.18
-5.41
0.18
a-hpa
-4.14
0.00
-3.02
0.32
-3.16
0.22
-6.03
0.22
a-hx2
-3.34
0.01
-2.74
0.20
-2.60
0.19
-4.79
0.18
a-hx3
-3.01
0.01
-2.39
0.25
-1.58
0.23
-3.94
0.23
a-mba
-1.76
0.02
-1.22
0.30
-0.89
0.25
-3.17
0.23
a-mha
-3.60
0.00
-3.60
0.29
-2.89
0.17
-5.55
0.22
a-mhp
-4.17
0.00
-3.98
0.29
-3.82
0.19
-6.23
0.21
a-nmb
-1.69
0.02
-1.95
0.42
-0.83
0.18
-2.74
0.21
a-nmh
-3.52
0.01
-4.15
0.59
-2.92
0.18
-5.56
0.19
a-oam
-4.61
0.01
-4.68
0.49
-4.33
0.17
-6.99
0.19
a-oct
-4.62
0.02
-4.64
0.30
-4.85
0.24
-6.81
0.19
a-pam
-2.72
0.00
-2.66
0.77
-1.53
0.18
-4.00
0.23
a-pnt
-2.60
0.01
-2.56
0.23
-1.74
0.19
-4.14
0.19
b-ben
-1.64
0.02
-2.85
0.62
-1.83
0.29
-2.45
0.17
b-cbu
-1.55
0.17
-1.88
0.20
-1.64
0.36
-2.77
0.17
b-chp
-4.56
0.01
-3.08
0.25
-2.79
0.34
-6.27
0.23
b-coc
-4.97
0.04
-3.28
0.23
-3.36
0.26
-7.13
0.22
b-cpe
-3.05
0.01
-3.57
0.34
-3.55
0.31
-5.93
0.27
b-ham
-2.49
0.08
-2.52
0.20
-2.01
0.26
-4.14
0.19
b-hep
-3.39
0.18
-3.41
0.28
-3.34
0.35
-4.15
0.23
b-hex
-2.28
0.03
-2.93
0.25
-2.47
0.27
-3.59
0.17
b-m4c
-4.32
0.01
-2.89
0.24
-2.68
0.29
-5.64
0.22
b-m4t
-4.54
0.01
-3.82
0.19
-3.50
0.26
-6.33
0.17
b-mch
-4.18
0.01
-3.69
0.22
-3.31
0.26
-6.07
0.17
b-mha
-2.56
0.07
-3.46
0.24
-2.14
0.28
-4.66
0.18
b-mo3
-2.16
0.01
-2.87
0.38
-2.73
0.41
-2.79
0.20
b-mo4
-2.51
0.01
-4.19
0.41
-3.10
0.31
-3.49
0.21
b-mp3
-1.46
0.04
-3.03
0.27
-2.48
0.28
-3.00
0.19
b-mp4
-2.19
0.01
-3.02
0.32
-2.77
0.34
-3.06
0.21
b-oam
-3.59
0.12
-3.35
0.28
-2.60
0.30
-5.25
0.23
b-pb3
-3.52
0.01
-3.49
0.32
-2.87
0.30
-4.58
0.17
b-pb4
-3.60
0.02
-3.62
0.33
-3.34
0.35
-4.71
0.23
b-pha
-1.70
0.05
-3.24
0.31
-2.55
0.29
-3.98
0.19
b-pnt
-1.27
0.32
-2.22
0.25
-1.73
0.29
-2.00
0.16
Table 6: Experimental and predicted binding enthalpies (ΔH). Values in kcal/mol.
System
Experimental
SMIRNOFF99Frosst
GAFF v1.7
GAFF v2.1
Mean
SEM
Mean
SEM
Mean
SEM
Mean
SEM
a-bam
-2.17
0.05
-0.43
0.28
-0.84
0.59
-3.05
0.38
a-but
-2.53
0.12
-0.76
0.59
-1.08
0.37
-4.91
0.42
a-cbu
-2.75
0.05
-2.08
0.21
-0.71
0.49
-4.94
0.29
a-chp
-2.99
0.23
-3.42
0.39
-2.33
0.28
-5.27
0.35
a-coc
-0.93
0.32
-3.80
0.45
-2.93
0.32
-6.17
0.32
a-cpe
-2.74
0.02
-1.93
0.30
-1.06
0.42
-4.86
0.29
a-ham
-4.19
0.02
-4.02
0.33
-2.33
0.30
-6.91
0.29
a-hep
-4.19
0.09
-4.72
0.33
-4.05
0.36
-8.68
0.24
a-hex
-3.40
0.02
-4.33
0.30
-2.95
0.30
-7.43
0.31
a-hp6
-4.48
0.02
-4.86
0.31
-3.73
0.21
-8.24
0.31
a-hpa
-4.66
0.02
-4.47
0.36
-2.65
0.30
-7.38
0.26
a-hx2
-4.12
0.06
-4.24
0.31
-2.35
0.32
-6.56
0.30
a-hx3
-3.36
0.05
-2.25
0.55
-2.80
0.32
-5.51
0.28
a-mba
-2.68
0.07
-0.95
0.41
-0.32
0.37
-3.11
0.36
a-mha
-4.28
0.02
-3.31
0.50
-2.16
0.25
-6.40
0.34
a-mhp
-4.74
0.02
-4.89
0.23
-3.41
0.23
-8.12
0.28
a-nmb
-2.57
0.06
-1.10
0.28
0.03
0.23
-3.34
0.27
a-nmh
-4.20
0.08
-4.20
0.48
-2.54
0.24
-6.74
0.30
a-oam
-5.46
0.03
-4.93
0.28
-3.73
0.33
-8.02
0.28
a-oct
-4.89
0.03
-6.08
0.21
-4.69
0.29
-9.53
0.30
a-pam
-3.28
0.02
-1.72
0.61
-0.84
0.28
-4.45
0.33
a-pnt
-2.75
0.01
-2.05
0.37
-1.62
0.33
-5.99
0.30
b-ben
-2.51
0.08
-0.45
0.56
-0.76
0.82
-1.30
0.26
b-cbu
0.88
0.17
0.05
0.83
-0.19
0.46
0.87
0.29
b-chp
-2.96
0.01
0.88
0.40
1.82
0.61
-4.39
0.31
b-coc
-3.92
0.06
0.80
0.47
0.45
0.98
-5.57
0.44
b-cpe
-1.09
0.01
1.86
0.37
3.62
0.65
-1.32
0.30
b-ham
0.60
0.05
1.66
0.68
2.29
0.78
0.42
0.33
b-hep
0.42
0.04
-0.05
0.38
1.91
0.29
-0.92
0.27
b-hex
1.31
0.04
0.41
0.65
1.30
0.60
-0.21
0.27
b-m4c
-2.27
0.01
2.18
0.48
2.62
0.55
-3.13
0.30
b-m4t
-2.17
0.02
1.26
0.45
2.49
0.51
-3.11
0.29
b-mch
-2.29
0.03
-0.79
1.08
2.27
0.73
-3.37
0.34
b-mha
0.47
0.03
0.53
0.55
2.48
0.64
0.28
0.29
b-mo3
-2.93
0.03
-2.66
0.46
-0.59
0.45
-2.50
0.28
b-mo4
-1.96
0.01
-2.69
0.58
-0.91
0.33
-3.05
0.29
b-mp3
-2.75
0.13
-1.09
0.68
0.68
0.48
-2.64
0.31
b-mp4
-2.89
0.05
-2.84
0.76
0.78
0.60
-2.34
0.28
b-oam
-0.48
0.03
0.98
0.41
2.66
0.43
-0.52
0.34
b-pb3
-2.25
0.01
-1.59
0.94
1.78
0.36
-2.24
0.30
b-pb4
-2.82
0.01
-0.02
0.62
-1.44
0.78
-3.70
0.29
b-pha
-1.79
0.11
-1.10
0.69
-1.34
0.97
-3.45
0.36
b-pnt
1.89
0.53
-0.79
1.01
-0.51
0.84
0.40
0.31
Table 7: Experimental and predicted binding entropies (−TΔS°). Values in kcal/mol.
System
Experimental
SMIRNOFF99Frosst
GAFF v1.7
GAFF v2.1
Mean
SEM
Mean
SEM
Mean
SEM
Mean
SEM
a-bam
0.59
0.05
-2.82
0.53
0.02
0.63
0.11
0.45
a-but
1.02
0.13
-0.73
0.65
-0.01
0.42
1.77
0.47
a-cbu
0.73
0.05
0.75
0.28
-0.17
0.54
1.21
0.36
a-chp
0.48
0.24
1.04
0.48
0.63
0.37
1.16
0.41
a-coc
-2.30
1.18
2.02
0.53
1.07
0.40
2.82
0.40
a-cpe
0.61
0.03
0.33
0.39
-0.44
0.51
1.07
0.37
a-ham
0.66
0.02
0.59
0.44
-0.68
0.36
0.92
0.34
a-hep
0.20
0.09
0.77
0.39
0.12
0.41
2.45
0.29
a-hex
0.02
0.02
1.62
0.37
0.03
0.36
2.17
0.37
a-hp6
0.88
0.02
1.54
0.38
0.37
0.27
2.84
0.36
a-hpa
0.52
0.02
1.45
0.48
-0.50
0.37
1.35
0.34
a-hx2
0.78
0.06
1.49
0.37
-0.25
0.37
1.77
0.35
a-hx3
0.35
0.05
-0.13
0.61
1.23
0.40
1.58
0.36
a-mba
0.92
0.07
-0.27
0.51
-0.57
0.44
-0.06
0.43
a-mha
0.68
0.02
-0.29
0.58
-0.73
0.30
0.85
0.40
a-mhp
0.57
0.02
0.92
0.37
-0.41
0.30
1.89
0.35
a-nmb
0.88
0.06
-0.85
0.50
-0.86
0.29
0.61
0.35
a-nmh
0.68
0.08
0.05
0.76
-0.38
0.30
1.18
0.36
a-oam
0.85
0.03
0.25
0.57
-0.60
0.37
1.02
0.34
a-oct
0.27
0.04
1.44
0.37
-0.16
0.38
2.72
0.36
a-pam
0.56
0.02
-0.94
0.98
-0.68
0.34
0.45
0.40
a-pnt
0.15
0.01
-0.51
0.43
-0.12
0.38
1.84
0.36
b-ben
0.87
0.08
-2.40
0.83
-1.07
0.87
-1.15
0.31
b-cbu
-2.43
0.24
-1.93
0.85
-1.45
0.58
-3.64
0.34
b-chp
-1.60
0.01
-3.96
0.47
-4.61
0.69
-1.87
0.39
b-coc
-1.05
0.07
-4.08
0.53
-3.81
1.02
-1.56
0.49
b-cpe
-1.96
0.01
-5.43
0.51
-7.17
0.72
-4.60
0.40
b-ham
-3.09
0.09
-4.19
0.71
-4.29
0.82
-4.56
0.38
b-hep
-3.81
0.18
-3.36
0.47
-5.25
0.45
-3.23
0.35
b-hex
-3.59
0.05
-3.34
0.70
-3.77
0.66
-3.38
0.32
b-m4c
-2.05
0.01
-5.07
0.54
-5.29
0.62
-2.51
0.37
b-m4t
-2.37
0.02
-5.08
0.49
-5.99
0.57
-3.22
0.33
b-mch
-1.89
0.03
-2.90
1.10
-5.58
0.78
-2.70
0.38
b-mha
-3.03
0.08
-4.00
0.60
-4.62
0.69
-4.94
0.34
b-mo3
0.77
0.03
-0.20
0.60
-2.14
0.61
-0.29
0.34
b-mo4
-0.55
0.01
-1.50
0.71
-2.19
0.46
-0.45
0.36
b-mp3
1.29
0.14
-1.94
0.74
-3.16
0.56
-0.35
0.37
b-mp4
0.70
0.05
-0.19
0.83
-3.55
0.69
-0.72
0.35
b-oam
-3.11
0.12
-4.33
0.50
-5.26
0.53
-4.73
0.41
b-pb3
-1.27
0.01
-1.90
0.99
-4.64
0.47
-2.34
0.34
b-pb4
-0.78
0.02
-3.60
0.71
-1.90
0.85
-1.01
0.37
b-pha
0.09
0.12
-2.14
0.76
-1.21
1.01
-0.52
0.41
b-pnt
-3.16
0.62
-1.43
1.04
-1.22
0.89
-2.40
0.35
Table 8: Predicted thermodynamic properties for SMIRNOFF99Frosst relative to experiment in kcal/mol, analyzed using MBAR.
RMSE
MSE
R²
Slope
Intercept
Tau
ΔG°
SMIRNOFF99Frosst
0.80
[0.62, 1.00]
-0.04
[-0.28, 0.20]
0.67
[0.22, 0.65]
0.53
[0.33, 0.73]
-1.45
[-0.80, -2.07]
0.48
[0.32, 0.62]
ΔH
SMIRNOFF99Frosst
1.83
[1.37, 2.28]
0.73
[0.26, 1.24]
0.44
[0.21, 0.66]
0.84
[0.54, 1.18]
0.36
[-0.53, 1.47]
0.52
[0.34, 0.68]
−TΔS°
SMIRNOFF99Frosst
1.84
[1.44, 2.25]
-0.76
[-1.26, -0.26]
0.40
[0.15, 0.63]
0.87
[0.50, 1.24]
-0.84
[-1.33, -0.36]
0.32
[0.11, -0.50]
Table 9: Predicted thermodynamic properties for each force field relative to experiment on ammonium guests. Values in kcal/mol.
RMSE
MSE
R²
Slope
Intercept
Tau
ΔG°
SMIRNOFF99Frosst
0.76
[0.43, 1.11]
-0.10
[-0.54, 0.31]
0.48
[0.07, 0.84]
0.69
[0.19, 1.16]
-1.06
[0.54, -2.77]
0.44
[0.75, 0.04]
ΔG°
GAFF v1.7
0.77
[0.59, 0.95]
0.69
[0.51, 0.88]
0.90
[0.76, 0.98]
1.08
[0.88, 1.26]
0.95
[1.56, 0.32]
0.74
[0.91, 0.50]
ΔG°
GAFF v2.1
1.85
[1.59, 2.09]
-1.79
[-2.04, -1.53]
0.93
[0.83, 0.98]
1.32
[1.13, 1.51]
-0.80
[-0.20, -1.46]
0.76
[0.92, 0.53]
ΔH
SMIRNOFF99Frosst
1.15
[0.77, 1.51]
0.83
[0.39, 1.27]
0.89
[0.76, 0.97]
1.15
[0.89, 1.53]
1.31
[2.81, 0.38]
0.78
[0.92, 0.56]
ΔH
GAFF v1.7
2.12
[1.77, 2.47]
2.02
[1.67, 2.37]
0.92
[0.80, 0.98]
1.09
[0.86, 1.35]
2.29
[3.34, 1.39]
0.75
[0.90, 0.54]
ΔH
GAFF v2.1
1.90
[1.31, 2.43]
-1.51
[-2.15, -0.88]
0.96
[0.91, 0.99]
1.54
[1.38, 1.83]
0.09
[1.18, -0.44]
0.81
[0.95, 0.62]
−TΔS°
SMIRNOFF99Frosst
1.47
[0.90, 2.10]
-0.93
[-1.59, -0.31]
0.65
[0.13, 0.91]
0.99
[0.58, 1.35]
-0.88
[-0.09, -1.66]
0.26
[0.64, -0.26]
−TΔS°
GAFF v1.7
1.45
[1.14, 1.79]
-1.33
[-1.66, -1.00]
0.88
[0.18, 0.97]
1.04
[-0.02, 1.37]
-1.27
[-0.55, -1.62]
0.28
[0.64, -0.21]
−TΔS°
GAFF v2.1
1.04
[0.67, 1.40]
-0.27
[-0.84, 0.26]
0.89
[0.29, 0.98]
1.36
[-0.53, 1.66]
-0.12
[1.16, -0.59]
0.23
[0.62, -0.26]
Table 10: Predicted thermodynamic properties for each force field relative to experiment on carboxylate guests. Values in kcal/mol.
RMSE
MSE
R²
Slope
Intercept
Tau
ΔG°
SMIRNOFF99Frosst
0.87
[0.59, 1.16]
-0.36
[-0.74, -0.01]
0.34
[0.02, 0.68]
0.45
[0.11, 0.75]
-1.85
[-0.91, -2.83]
0.40
[0.67, 0.07]
ΔG°
GAFF v1.7
0.68
[0.49, 0.88]
0.03
[-0.28, 0.34]
0.52
[0.16, 0.80]
0.68
[0.33, 0.97]
-0.84
[0.08, -1.75]
0.53
[0.76, 0.23]
ΔG°
GAFF v2.1
1.46
[1.21, 1.71]
-1.36
[-1.61, -1.10]
0.81
[0.61, 0.93]
1.18
[0.85, 1.46]
-0.87
[0.02, -1.74]
0.72
[0.87, 0.54]
ΔH
SMIRNOFF99Frosst
1.41
[0.94, 1.93]
0.20
[-0.43, 0.84]
0.53
[0.20, 0.79]
0.83
[0.40, 1.53]
-0.14
[2.12, -1.30]
0.59
[0.80, 0.30]
ΔH
GAFF v1.7
1.95
[1.34, 2.55]
1.24
[0.55, 1.93]
0.47
[0.13, 0.78]
0.79
[0.32, 1.49]
0.82
[3.10, -0.54]
0.53
[0.75, 0.23]
ΔH
GAFF v2.1
2.43
[1.75, 3.06]
-1.73
[-2.51, -0.96]
0.69
[0.49, 0.85]
1.40
[0.99, 2.29]
-0.66
[2.15, -1.61]
0.63
[0.82, 0.35]
−TΔS°
SMIRNOFF99Frosst
1.73
[1.17, 2.29]
-0.57
[-1.32, 0.16]
0.29
[0.02, 0.61]
0.62
[0.16, 1.09]
-0.68
[0.05, -1.43]
0.27
[0.58, -0.09]
−TΔS°
GAFF v1.7
2.07
[1.35, 2.76]
-1.22
[-2.00, -0.46]
0.29
[0.00, 0.67]
0.63
[-0.02, 1.18]
-1.31
[-0.58, -2.09]
0.27
[0.58, -0.09]
−TΔS°
GAFF v2.1
1.46
[1.12, 1.77]
0.37
[-0.27, 1.00]
0.50
[0.13, 0.76]
0.93
[0.58, 1.30]
0.37
[1.07, -0.34]
0.37
[0.67, -0.01]
Table 11: Predicted thermodynamic properties for each force field relative to experiment on cyclic alcohol guests. Values in kcal/mol.
RMSE
MSE
R²
Slope
Intercept
Tau
ΔG°
SMIRNOFF99Frosst
1.07
[0.66, 1.58]
0.71
[0.22, 1.21]
0.54
[0.09, 0.86]
0.55
[0.20, 0.84]
-0.84
[0.16, -2.09]
0.44
[0.75, 0.02]
ΔG°
GAFF v1.7
1.22
[0.86, 1.67]
0.93
[0.45, 1.41]
0.56
[0.12, 0.89]
0.59
[0.25, 0.89]
-0.47
[0.64, -1.77]
0.47
[0.78, 0.05]
ΔG°
GAFF v2.1
1.80
[1.48, 2.15]
-1.64
[-2.04, -1.14]
0.73
[0.19, 0.98]
1.01
[0.49, 1.27]
-1.63
[-0.67, -3.19]
0.66
[0.89, 0.27]
ΔH
SMIRNOFF99Frosst
2.88
[1.99, 3.68]
1.66
[0.21, 3.03]
0.09
[0.00, 0.44]
0.07
[-1.28, 1.67]
-0.29
[3.93, -4.06]
0.09
[0.56, -0.35]
ΔH
GAFF v1.7
3.63
[2.67, 4.47]
2.66
[1.13, 4.07]
0.10
[0.00, 0.57]
0.12
[-1.09, 2.28]
0.91
[6.66, -2.47]
0.14
[0.60, -0.31]
ΔH
GAFF v2.1
2.08
[1.18, 3.16]
-1.64
[-2.54, -0.91]
0.54
[0.00, 0.97]
1.08
[-0.37, 1.90]
-1.51
[0.83, -5.50]
0.46
[0.89, -0.09]
−TΔS°
SMIRNOFF99Frosst
2.47
[1.62, 3.36]
-0.96
[-2.22, 0.52]
0.40
[0.00, 0.93]
1.18
[-0.45, 2.26]
-0.88
[0.36, -3.60]
0.30
[0.71, -0.20]
−TΔS°
GAFF v1.7
3.00
[2.07, 3.88]
-1.73
[-3.14, -0.18]
0.37
[0.00, 0.93]
1.23
[-0.38, 2.48]
-1.59
[-0.31, -4.23]
0.29
[0.71, -0.20]
−TΔS°
GAFF v2.1
1.80
[0.68, 3.19]
-0.00
[-0.98, 1.27]
0.48
[0.00, 0.97]
1.13
[-0.22, 1.96]
0.08
[1.14, -1.79]
0.46
[0.82, -0.02]
Table 12: Dihedral parameter differences between SMIRNOFF99Frosst and GAFF v1.7. Atom names refer to Figure 2. NP: not present.
SMIRNOFF99Frosst
GAFF v1.7
Atom 1
Atom 2
Atom 3
Atom 4
Per
Phase
Height (kcal/mol)
Height (kcal/mol)
H1
C1
C2
O2
1
0
0.25
NP
H1
C1
C2
O2
3
0
0.00
0.16
Table 13: Dihedral parameter differences between SMIRNOFF99Frosst and GAFF v2.1, where one dihedral has fewer or more periodicity terms than the corresponding term in the other force field. Atom names refer to 2. NP: not present.
 0000-0003-3928-5050 ·
0000-0003-3928-5050 ·  slochower ·
slochower ·  drslochower
drslochower